
Upload README.md
Browse files
README.md
CHANGED
|
@@ -1,39 +1,42 @@
|
|
| 1 |
---
|
| 2 |
-
|
| 3 |
-
|
| 4 |
-
|
| 5 |
-
|
| 6 |
-
- name: unaligned_seqs
|
| 7 |
-
struct:
|
| 8 |
-
- name: seq0
|
| 9 |
-
dtype: string
|
| 10 |
-
- name: seq1
|
| 11 |
-
dtype: string
|
| 12 |
-
- name: seq2
|
| 13 |
-
dtype: string
|
| 14 |
-
- name: seq3
|
| 15 |
-
dtype: string
|
| 16 |
-
- name: seq4
|
| 17 |
-
dtype: string
|
| 18 |
-
- name: seq5
|
| 19 |
-
dtype: string
|
| 20 |
-
- name: seq6
|
| 21 |
-
dtype: string
|
| 22 |
-
- name: seq7
|
| 23 |
-
dtype: string
|
| 24 |
-
splits:
|
| 25 |
-
- name: train
|
| 26 |
-
num_bytes: 1052034201
|
| 27 |
-
num_examples: 1494999
|
| 28 |
-
- name: validation
|
| 29 |
-
num_bytes: 1413489
|
| 30 |
-
num_examples: 2000
|
| 31 |
-
- name: test
|
| 32 |
-
num_bytes: 2110321
|
| 33 |
-
num_examples: 3001
|
| 34 |
-
download_size: 494773954
|
| 35 |
-
dataset_size: 1055558011
|
| 36 |
---
|
| 37 |
-
#
|
|
|
|
|
|
|
|
|
|
| 38 |
|
| 39 |
-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
---
|
| 2 |
+
tags:
|
| 3 |
+
- sequence-to-sequence
|
| 4 |
+
- bioinformatics
|
| 5 |
+
- biology
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 6 |
---
|
| 7 |
+
# Multiple Sequence Alignment as a Sequence-to-Sequence Learning Problem
|
| 8 |
+
## Abstract:
|
| 9 |
+
The sequence alignment problem is one of the most fundamental problems in bioinformatics and a plethora of methods were devised to tackle it. Here we introduce BetaAlign, a methodology for aligning sequences using an NLP approach. BetaAlign accounts for the possible variability of the evolutionary process among different datasets by using an ensemble of transformers, each trained on millions of samples generated from a different evolutionary model. Our approach leads to alignment accuracy that is similar and often better than commonly used methods, such as MAFFT, DIALIGN, ClustalW, T-Coffee, PRANK, and MUSCLE.
|
| 10 |
+
|
| 11 |
|
| 12 |
+
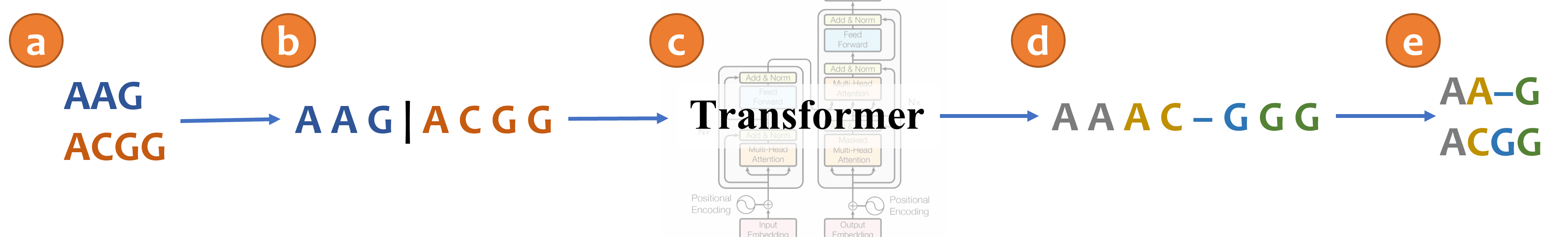
An illustration of aligning sequences with sequence-to-sequence learning. (a) Consider two input sequences "AAG" and "ACGG". (b) The result of encoding the unaligned sequences into the source language (*Concat* representation). (c) The sentence from the source language is translated to the target language via a transformer model. (d) The translated sentence in the target language (*Spaces* representation). (e) The resulting alignment, decoded from the translated sentence, in which "AA-G" is aligned to "ACGG". The transformer architecture illustration is adapted from (Vaswani et al., 2017).
|
| 13 |
+
|
| 14 |
+
## Data:
|
| 15 |
+
|
| 16 |
+
We used SpartaABC (Loewenthal et al., 2021) to generate millions of true alignments. SpartaABC requires the following input: (1) a rooted phylogenetic tree, which includes a topology and branch lengths; (2) a substitution model (amino acids or nucleotides); (3) root sequence length; (4) the indel model parameters, which include: insertion rate (*R_I*), deletion rate (*R_D*), a parameter for the insertion Zipfian distribution (*A_I*), and a parameter for the deletion Zipfian distribution (*A_D*). MSAs were simulated along random phylogenetic tree topologies generated using the program ETE version 3.0 (Huerta-Cepas et al., 2016) with default parameters.
|
| 17 |
+
|
| 18 |
+
We generated 1,495,000, 2,000 and 3,000, protein MSAs with ten sequences that were used as training validation and testing data, respectively. We generated the same number of DNA MSAs. For each random tree, branch lengths were drawn from a uniform distribution in the range *(0.5,1.0)*. Next, the sequences were generated using SpartaABC with the following parameters: *R_I,R_D \in (0.0,0.05)*, *A_I, A_D \in (1.01,2.0)*. The alignment lengths as well as the sequence lengths of the tree leaves vary within and among datasets as they depend on the indel dynamics and the root length. The root length was sampled uniformly in the range *[32,44]*. Unless stated otherwise, all protein datasets were generated with the WAG+G model, and all DNA datasets were generated with the GTR+G model, with the following parameters: (1) frequencies for the different nucleotides *(0.37, 0.166, 0.307, 0.158)*, in the order "T", "C", "A" and "G"; (2) with the substitutions rate *(0.444, 0.0843, 0.116, 0.107, 0.00027)*, in the order "a", "b", "c", "d", and "e" for the substitution matrix.
|
| 19 |
+
|
| 20 |
+
## Example:
|
| 21 |
+
|
| 22 |
+
The following example correspond for the illustrated MSA in the figure above:
|
| 23 |
+
|
| 24 |
+
{"MSA": "AAAC-GGG", "unaligned_seqs": {"seq0": "AAG", "seq1": "ACGG"}}
|
| 25 |
+
|
| 26 |
+
## APA
|
| 27 |
+
|
| 28 |
+
```
|
| 29 |
+
Dotan, E., Belinkov, Y., Avram, O., Wygoda, E., Ecker, N., Alburquerque, M., Keren, O., Loewenthal, G., & Pupko T. (2023). Multiple sequence alignment as a sequence-to-sequence learning problem. The Eleventh International Conference on Learning Representations (ICLR 2023).
|
| 30 |
+
```
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
## BibTeX
|
| 34 |
+
```
|
| 35 |
+
@article{Dotan_multiple_2023,
|
| 36 |
+
author = {Dotan, Edo and Belinkov, Yonatan and Avram, Oren and Wygoda, Elya and Ecker, Noa and Alburquerque, Michael and Keren, Omri and Loewenthal, Gil and Pupko, Tal},
|
| 37 |
+
month = aug,
|
| 38 |
+
title = {{Multiple sequence alignment as a sequence-to-sequence learning problem}},
|
| 39 |
+
year = {2023}
|
| 40 |
+
}
|
| 41 |
+
|
| 42 |
+
```
|