Spaces:
Sleeping

Sleeping
Upload 46 files
Browse filesThis is the first raw knowledge base of OmicVerse
- ovrawm/t_anno_trans.txt +171 -0
- ovrawm/t_aucell.txt +93 -0
- ovrawm/t_bulk_combat.txt +205 -0
- ovrawm/t_cellanno.txt +378 -0
- ovrawm/t_cellfate.txt +218 -0
- ovrawm/t_cellfate_gene.txt +468 -0
- ovrawm/t_cellfate_genesets.txt +181 -0
- ovrawm/t_cellphonedb.txt +439 -0
- ovrawm/t_cluster.txt +312 -0
- ovrawm/t_cluster_space.txt +399 -0
- ovrawm/t_cnmf.txt +331 -0
- ovrawm/t_commot_flowsig.txt +395 -0
- ovrawm/t_cytotrace.txt +110 -0
- ovrawm/t_deg.txt +323 -0
- ovrawm/t_deseq2.txt +237 -0
- ovrawm/t_gptanno.txt +330 -0
- ovrawm/t_mapping.txt +191 -0
- ovrawm/t_metacells.txt +249 -0
- ovrawm/t_metatime.txt +177 -0
- ovrawm/t_mofa.txt +184 -0
- ovrawm/t_mofa_glue.txt +255 -0
- ovrawm/t_network.txt +88 -0
- ovrawm/t_nocd.txt +112 -0
- ovrawm/t_preprocess.txt +421 -0
- ovrawm/t_preprocess_cpu.txt +404 -0
- ovrawm/t_preprocess_gpu.txt +416 -0
- ovrawm/t_scdeg.txt +316 -0
- ovrawm/t_scdrug.txt +225 -0
- ovrawm/t_scmulan.txt +199 -0
- ovrawm/t_simba.txt +146 -0
- ovrawm/t_single_batch.txt +333 -0
- ovrawm/t_slat.txt +365 -0
- ovrawm/t_spaceflow.txt +160 -0
- ovrawm/t_stagate.txt +296 -0
- ovrawm/t_staligner.txt +155 -0
- ovrawm/t_starfysh.txt +519 -0
- ovrawm/t_stt.txt +274 -0
- ovrawm/t_tcga.txt +96 -0
- ovrawm/t_tosica.txt +317 -0
- ovrawm/t_traj.txt +238 -0
- ovrawm/t_via.txt +193 -0
- ovrawm/t_via_velo.txt +102 -0
- ovrawm/t_visualize_bulk.txt +170 -0
- ovrawm/t_visualize_colorsystem.txt +223 -0
- ovrawm/t_visualize_single.txt +534 -0
- ovrawm/t_wgcna.txt +252 -0
ovrawm/t_anno_trans.txt
ADDED
|
@@ -0,0 +1,171 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Celltype annotation transfer in multi-omics
|
| 5 |
+
#
|
| 6 |
+
# In the field of multi-omics research, transferring cell type annotations from one data modality to another is a crucial step. For instance, when annotating cell types in single-cell ATAC sequencing (scATAC-seq) data, it's often desirable to leverage the cell type labels already annotated in single-cell RNA sequencing (scRNA-seq) data. This process involves integrating information from both scRNA-seq and scATAC-seq data modalities.
|
| 7 |
+
#
|
| 8 |
+
# GLUE is a prominent algorithm used for cross-modality integration, allowing researchers to combine data from different omics modalities effectively. However, GLUE does not inherently provide a method for transferring cell type labels from scRNA-seq to scATAC-seq data. To address this limitation, an approach was implemented in the omicverse platform using K-nearest neighbor (KNN) graphs.
|
| 9 |
+
#
|
| 10 |
+
# The KNN graph-based approach likely involves constructing KNN graphs separately for scRNA-seq and scATAC-seq data. In these graphs, each cell is connected to its K nearest neighbors based on certain similarity metrics, which could be calculated using gene expression profiles in scRNA-seq and accessibility profiles in scATAC-seq. Once these graphs are constructed, the idea is to transfer the cell type labels from the scRNA-seq side to the scATAC-seq side by assigning labels to scATAC-seq cells based on the labels of their KNN neighbors in the scRNA-seq graph.
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1aIMmSgyIw-PGjJ65WvMgz4Ob3EtoK_UV?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[3]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
import matplotlib.pyplot as plt
|
| 19 |
+
import scanpy as sc
|
| 20 |
+
ov.ov_plot_set()
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
# ## Loading the data preprocessed with GLUE
|
| 24 |
+
#
|
| 25 |
+
# Here, we use two output files from the GLUE cross-modal integration, and their common feature is that they both have the `obsm['X_glue']` layer. And the rna have been annotated.
|
| 26 |
+
|
| 27 |
+
# In[4]:
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
rna=sc.read("data/analysis_lymph/rna-emb.h5ad")
|
| 31 |
+
atac=sc.read("data/analysis_lymph/atac-emb.h5ad")
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# We can visualize the intergrated effect of GLUE with UMAP
|
| 35 |
+
|
| 36 |
+
# In[5]:
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
import scanpy as sc
|
| 40 |
+
combined=sc.concat([rna,atac],merge='same')
|
| 41 |
+
combined
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# In[6]:
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
combined.obsm['X_mde']=ov.utils.mde(combined.obsm['X_glue'])
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
# We can see that the two layers are correctly aligned
|
| 51 |
+
|
| 52 |
+
# In[8]:
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
ov.utils.embedding(combined,
|
| 56 |
+
basis='X_mde',
|
| 57 |
+
color='domain',
|
| 58 |
+
title='Layers',
|
| 59 |
+
show=False,
|
| 60 |
+
palette=ov.utils.red_color,
|
| 61 |
+
frameon='small'
|
| 62 |
+
)
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
# And the RNA modality has an already annotated cell type label on it
|
| 66 |
+
|
| 67 |
+
# In[22]:
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
ov.utils.embedding(rna,
|
| 71 |
+
basis='X_mde',
|
| 72 |
+
color='major_celltype',
|
| 73 |
+
title='Cell type',
|
| 74 |
+
show=False,
|
| 75 |
+
#palette=ov.utils.red_color,
|
| 76 |
+
frameon='small'
|
| 77 |
+
)
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
# ## Celltype transfer
|
| 81 |
+
#
|
| 82 |
+
# We train a knn nearest neighbour classifier using `X_glue` features
|
| 83 |
+
|
| 84 |
+
# In[13]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
knn_transformer=ov.utils.weighted_knn_trainer(
|
| 88 |
+
train_adata=rna,
|
| 89 |
+
train_adata_emb='X_glue',
|
| 90 |
+
n_neighbors=15,
|
| 91 |
+
)
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
# In[14]:
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
labels,uncert=ov.utils.weighted_knn_transfer(
|
| 98 |
+
query_adata=atac,
|
| 99 |
+
query_adata_emb='X_glue',
|
| 100 |
+
label_keys='major_celltype',
|
| 101 |
+
knn_model=knn_transformer,
|
| 102 |
+
ref_adata_obs=rna.obs,
|
| 103 |
+
)
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
# We migrate the training results of the KNN classifier to atac. `unc` stands for uncertainty, with higher uncertainty demonstrating lower migration accuracy, suggesting that the cell in question may be a double-fate signature or some other type of cell.
|
| 107 |
+
|
| 108 |
+
# In[15]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
atac.obs["transf_celltype"]=labels.loc[atac.obs.index,"major_celltype"]
|
| 112 |
+
atac.obs["transf_celltype_unc"]=uncert.loc[atac.obs.index,"major_celltype"]
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# In[24]:
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
atac.obs["major_celltype"]=atac.obs["transf_celltype"].copy()
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# In[27]:
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
ov.utils.embedding(atac,
|
| 125 |
+
basis='X_umap',
|
| 126 |
+
color=['transf_celltype_unc','transf_celltype'],
|
| 127 |
+
#title='Cell type Un',
|
| 128 |
+
show=False,
|
| 129 |
+
palette=ov.palette()[11:],
|
| 130 |
+
frameon='small'
|
| 131 |
+
)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# ## Visualization
|
| 135 |
+
#
|
| 136 |
+
# We can merge atac and rna after migration annotation and observe on the umap plot whether the cell types are consistent after merging the modalities.
|
| 137 |
+
|
| 138 |
+
# In[28]:
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
import scanpy as sc
|
| 142 |
+
combined1=sc.concat([rna,atac],merge='same')
|
| 143 |
+
combined1
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
# In[29]:
|
| 147 |
+
|
| 148 |
+
|
| 149 |
+
combined1.obsm['X_mde']=ov.utils.mde(combined1.obsm['X_glue'])
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
# We found that the annotation was better, suggesting that the KNN nearest-neighbour classifier we constructed can effectively migrate cell type labels from RNA to ATAC.
|
| 153 |
+
|
| 154 |
+
# In[31]:
|
| 155 |
+
|
| 156 |
+
|
| 157 |
+
ov.utils.embedding(combined1,
|
| 158 |
+
basis='X_mde',
|
| 159 |
+
color=['domain','major_celltype'],
|
| 160 |
+
title=['Layers','Cell type'],
|
| 161 |
+
show=False,
|
| 162 |
+
palette=ov.palette()[11:],
|
| 163 |
+
frameon='small'
|
| 164 |
+
)
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# In[ ]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
|
ovrawm/t_aucell.txt
ADDED
|
@@ -0,0 +1,93 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
import omicverse as ov
|
| 2 |
+
import scanpy as sc
|
| 3 |
+
import scvelo as scv
|
| 4 |
+
|
| 5 |
+
ov.utils.ov_plot_set()
|
| 6 |
+
|
| 7 |
+
ov.utils.download_pathway_database()
|
| 8 |
+
ov.utils.download_geneid_annotation_pair()
|
| 9 |
+
|
| 10 |
+
adata = scv.datasets.pancreas()
|
| 11 |
+
adata
|
| 12 |
+
|
| 13 |
+
adata.X.max()
|
| 14 |
+
|
| 15 |
+
sc.pp.normalize_total(adata, target_sum=1e4)
|
| 16 |
+
sc.pp.log1p(adata)
|
| 17 |
+
|
| 18 |
+
adata.X.max()
|
| 19 |
+
|
| 20 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Biological_Process_2021.txt',organism='Mouse')
|
| 21 |
+
|
| 22 |
+
##Assest one geneset
|
| 23 |
+
geneset_name='response to vitamin (GO:0033273)'
|
| 24 |
+
ov.single.geneset_aucell(adata,
|
| 25 |
+
geneset_name=geneset_name,
|
| 26 |
+
geneset=pathway_dict[geneset_name])
|
| 27 |
+
sc.pl.embedding(adata,
|
| 28 |
+
basis='umap',
|
| 29 |
+
color=["{}_aucell".format(geneset_name)])
|
| 30 |
+
|
| 31 |
+
##Assest more than one geneset
|
| 32 |
+
geneset_names=['response to vitamin (GO:0033273)','response to vitamin D (GO:0033280)']
|
| 33 |
+
ov.single.pathway_aucell(adata,
|
| 34 |
+
pathway_names=geneset_names,
|
| 35 |
+
pathways_dict=pathway_dict)
|
| 36 |
+
sc.pl.embedding(adata,
|
| 37 |
+
basis='umap',
|
| 38 |
+
color=[i+'_aucell' for i in geneset_names])
|
| 39 |
+
|
| 40 |
+
##Assest test geneset
|
| 41 |
+
ov.single.geneset_aucell(adata,
|
| 42 |
+
geneset_name='Sox',
|
| 43 |
+
geneset=['Sox17', 'Sox4', 'Sox7', 'Sox18', 'Sox5'])
|
| 44 |
+
sc.pl.embedding(adata,
|
| 45 |
+
basis='umap',
|
| 46 |
+
color=["Sox_aucell"])
|
| 47 |
+
|
| 48 |
+
##Assest all pathways
|
| 49 |
+
adata_aucs=ov.single.pathway_aucell_enrichment(adata,
|
| 50 |
+
pathways_dict=pathway_dict,
|
| 51 |
+
num_workers=8)
|
| 52 |
+
|
| 53 |
+
adata_aucs.obs=adata[adata_aucs.obs.index].obs
|
| 54 |
+
adata_aucs.obsm=adata[adata_aucs.obs.index].obsm
|
| 55 |
+
adata_aucs.obsp=adata[adata_aucs.obs.index].obsp
|
| 56 |
+
adata_aucs
|
| 57 |
+
|
| 58 |
+
adata_aucs.write_h5ad('data/pancreas_auce.h5ad',compression='gzip')
|
| 59 |
+
|
| 60 |
+
adata_aucs=sc.read('data/pancreas_auce.h5ad')
|
| 61 |
+
|
| 62 |
+
sc.pl.embedding(adata_aucs,
|
| 63 |
+
basis='umap',
|
| 64 |
+
color=geneset_names)
|
| 65 |
+
|
| 66 |
+
#adata_aucs.uns['log1p']['base']=None
|
| 67 |
+
sc.tl.rank_genes_groups(adata_aucs, 'clusters', method='t-test',n_genes=100)
|
| 68 |
+
sc.pl.rank_genes_groups_dotplot(adata_aucs,groupby='clusters',
|
| 69 |
+
cmap='Spectral_r',
|
| 70 |
+
standard_scale='var',n_genes=3)
|
| 71 |
+
|
| 72 |
+
degs = sc.get.rank_genes_groups_df(adata_aucs, group='Beta', key='rank_genes_groups', log2fc_min=2,
|
| 73 |
+
pval_cutoff=0.05)['names'].squeeze()
|
| 74 |
+
degs
|
| 75 |
+
|
| 76 |
+
import matplotlib.pyplot as plt
|
| 77 |
+
#fig, axes = plt.subplots(4,3,figsize=(12,9))
|
| 78 |
+
axes=sc.pl.embedding(adata_aucs,ncols=3,
|
| 79 |
+
basis='umap',show=False,return_fig=True,wspace=0.55,hspace=0.65,
|
| 80 |
+
color=['clusters']+degs.values.tolist(),
|
| 81 |
+
title=[ov.utils.plot_text_set(i,3,20)for i in ['clusters']+degs.values.tolist()])
|
| 82 |
+
|
| 83 |
+
axes.tight_layout()
|
| 84 |
+
|
| 85 |
+
adata.uns['log1p']['base']=None
|
| 86 |
+
sc.tl.rank_genes_groups(adata, 'clusters', method='t-test',n_genes=100)
|
| 87 |
+
|
| 88 |
+
res=ov.single.pathway_enrichment(adata,pathways_dict=pathway_dict,organism='Mouse',
|
| 89 |
+
group_by='clusters',plot=True)
|
| 90 |
+
|
| 91 |
+
ax=ov.single.pathway_enrichment_plot(res,plot_title='Enrichment',cmap='Reds',
|
| 92 |
+
xticklabels=True,cbar=False,square=True,vmax=10,
|
| 93 |
+
yticklabels=True,cbar_kws={'label': '-log10(qvalue)','shrink': 0.5,})
|
ovrawm/t_bulk_combat.txt
ADDED
|
@@ -0,0 +1,205 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Batch correction in Bulk RNA-seq or microarray data
|
| 5 |
+
#
|
| 6 |
+
# Variability in datasets are not only the product of biological processes: they are also the product of technical biases (Lander et al, 1999). ComBat is one of the most widely used tool for correcting those technical biases called batch effects.
|
| 7 |
+
#
|
| 8 |
+
# pyComBat (Behdenna et al, 2020) is a new Python implementation of ComBat (Johnson et al, 2007), a software widely used for the adjustment of batch effects in microarray data. While the mathematical framework is strictly the same, pyComBat:
|
| 9 |
+
#
|
| 10 |
+
# - has similar results in terms of batch effects correction;
|
| 11 |
+
# - is as fast or faster than the R implementation of ComBat and;
|
| 12 |
+
# - offers new tools for the community to participate in its development.
|
| 13 |
+
#
|
| 14 |
+
# Paper: [pyComBat, a Python tool for batch effects correction in high-throughput molecular data using empirical Bayes methods](https://doi.org/10.1101/2020.03.17.995431)
|
| 15 |
+
#
|
| 16 |
+
# Code: https://github.com/epigenelabs/pyComBat
|
| 17 |
+
#
|
| 18 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/121bbIiI3j4pTZ3yA_5p8BRkRyGMMmNAq?usp=sharing
|
| 19 |
+
|
| 20 |
+
# In[7]:
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
import anndata
|
| 24 |
+
import pandas as pd
|
| 25 |
+
import omicverse as ov
|
| 26 |
+
ov.ov_plot_set()
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
# ## Loading dataset
|
| 30 |
+
#
|
| 31 |
+
# This minimal usage example illustrates how to use pyComBat in a default setting, and shows some results on ovarian cancer data, freely available on NCBI’s [Gene Expression Omnibus](https://www.ncbi.nlm.nih.gov/geo/), namely:
|
| 32 |
+
#
|
| 33 |
+
# - GSE18520
|
| 34 |
+
# - GSE66957
|
| 35 |
+
# - GSE69428
|
| 36 |
+
#
|
| 37 |
+
# The corresponding expression files are available on [GitHub](https://github.com/epigenelabs/pyComBat/tree/master/data).
|
| 38 |
+
|
| 39 |
+
# In[15]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
dataset_1 = pd.read_pickle("data/combat/GSE18520.pickle")
|
| 43 |
+
adata1=anndata.AnnData(dataset_1.T)
|
| 44 |
+
adata1.obs['batch']='1'
|
| 45 |
+
adata1
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# In[16]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
dataset_2 = pd.read_pickle("data/combat/GSE66957.pickle")
|
| 52 |
+
adata2=anndata.AnnData(dataset_2.T)
|
| 53 |
+
adata2.obs['batch']='2'
|
| 54 |
+
adata2
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
# In[17]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
dataset_3 = pd.read_pickle("data/combat/GSE69428.pickle")
|
| 61 |
+
adata3=anndata.AnnData(dataset_3.T)
|
| 62 |
+
adata3.obs['batch']='3'
|
| 63 |
+
adata3
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
# We use the concat function to join the three datasets together and take the intersection for the same genes
|
| 67 |
+
|
| 68 |
+
# In[18]:
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
adata=anndata.concat([adata1,adata2,adata3],merge='same')
|
| 72 |
+
adata
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
# ## Removing batch effect
|
| 76 |
+
|
| 77 |
+
# In[31]:
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
ov.bulk.batch_correction(adata,batch_key='batch')
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
# ## Saving results
|
| 84 |
+
#
|
| 85 |
+
# Raw datasets
|
| 86 |
+
|
| 87 |
+
# In[70]:
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
raw_data=adata.to_df().T
|
| 91 |
+
raw_data.head()
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
# Removing Batch datasets
|
| 95 |
+
|
| 96 |
+
# In[71]:
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
removing_data=adata.to_df(layer='batch_correction').T
|
| 100 |
+
removing_data.head()
|
| 101 |
+
|
| 102 |
+
|
| 103 |
+
# save
|
| 104 |
+
|
| 105 |
+
# In[ ]:
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
raw_data.to_csv('raw_data.csv')
|
| 109 |
+
removing_data.to_csv('removing_data.csv')
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
# You can also save adata object
|
| 113 |
+
|
| 114 |
+
# In[ ]:
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
adata.write_h5ad('adata_batch.h5ad',compression='gzip')
|
| 118 |
+
#adata=ov.read('adata_batch.h5ad')
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# ## Compare the dataset before and after correction
|
| 122 |
+
#
|
| 123 |
+
# We specify three different colours for three different datasets
|
| 124 |
+
|
| 125 |
+
# In[51]:
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
color_dict={
|
| 129 |
+
'1':ov.utils.red_color[1],
|
| 130 |
+
'2':ov.utils.blue_color[1],
|
| 131 |
+
'3':ov.utils.green_color[1],
|
| 132 |
+
}
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
# In[57]:
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
fig,ax=plt.subplots( figsize = (20,4))
|
| 139 |
+
bp=plt.boxplot(adata.to_df().T,patch_artist=True)
|
| 140 |
+
for i,batch in zip(range(adata.shape[0]),adata.obs['batch']):
|
| 141 |
+
bp['boxes'][i].set_facecolor(color_dict[batch])
|
| 142 |
+
ax.axis(False)
|
| 143 |
+
plt.show()
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
# In[58]:
|
| 147 |
+
|
| 148 |
+
|
| 149 |
+
fig,ax=plt.subplots( figsize = (20,4))
|
| 150 |
+
bp=plt.boxplot(adata.to_df(layer='batch_correction').T,patch_artist=True)
|
| 151 |
+
for i,batch in zip(range(adata.shape[0]),adata.obs['batch']):
|
| 152 |
+
bp['boxes'][i].set_facecolor(color_dict[batch])
|
| 153 |
+
ax.axis(False)
|
| 154 |
+
plt.show()
|
| 155 |
+
|
| 156 |
+
|
| 157 |
+
# In addition to using boxplots to observe the effect of batch removal, we can also use PCA to observe the effect of batch removal
|
| 158 |
+
|
| 159 |
+
# In[59]:
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
adata.layers['raw']=adata.X.copy()
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
# We first calculate the PCA on the raw dataset
|
| 166 |
+
|
| 167 |
+
# In[60]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
ov.pp.pca(adata,layer='raw',n_pcs=50)
|
| 171 |
+
adata
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# We then calculate the PCA on the batch_correction dataset
|
| 175 |
+
|
| 176 |
+
# In[61]:
|
| 177 |
+
|
| 178 |
+
|
| 179 |
+
ov.pp.pca(adata,layer='batch_correction',n_pcs=50)
|
| 180 |
+
adata
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
# In[62]:
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
ov.utils.embedding(adata,
|
| 187 |
+
basis='raw|original|X_pca',
|
| 188 |
+
color='batch',
|
| 189 |
+
frameon='small')
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
# In[63]:
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
ov.utils.embedding(adata,
|
| 196 |
+
basis='batch_correction|original|X_pca',
|
| 197 |
+
color='batch',
|
| 198 |
+
frameon='small')
|
| 199 |
+
|
| 200 |
+
|
| 201 |
+
# In[ ]:
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
|
ovrawm/t_cellanno.txt
ADDED
|
@@ -0,0 +1,378 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Celltype auto annotation with SCSA
|
| 5 |
+
# Single-cell transcriptomics allows the analysis of thousands of cells in a single experiment and the identification of novel cell types, states and dynamics in a variety of tissues and organisms. Standard experimental protocols and analytical workflows have been developed to create single-cell transcriptomic maps from tissues.
|
| 6 |
+
#
|
| 7 |
+
# This tutorial focuses on how to interpret this data to identify cell types, states, and other biologically relevant patterns with the goal of creating annotated cell maps.
|
| 8 |
+
#
|
| 9 |
+
# Paper: [SCSA: A Cell Type Annotation Tool for Single-Cell RNA-seq Data](https://doi.org/10.3389/fgene.2020.00490)
|
| 10 |
+
#
|
| 11 |
+
# Code: https://github.com/bioinfo-ibms-pumc/SCSA
|
| 12 |
+
#
|
| 13 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1BC6hPS0CyBhNu0BYk8evu57-ua1bAS0T?usp=sharing
|
| 14 |
+
#
|
| 15 |
+
# <div class="admonition warning">
|
| 16 |
+
# <p class="admonition-title">Note</p>
|
| 17 |
+
# <p>
|
| 18 |
+
# The annotation with SCSA can't be used in rare celltype annotations
|
| 19 |
+
# </p>
|
| 20 |
+
# </div>
|
| 21 |
+
#
|
| 22 |
+
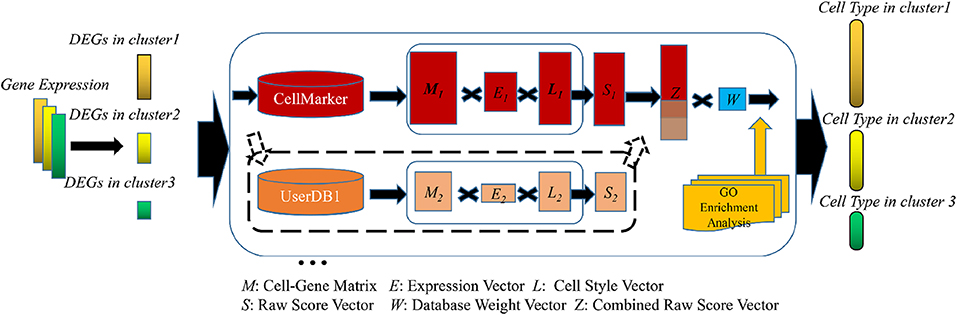
# 
|
| 23 |
+
|
| 24 |
+
# In[1]:
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
import omicverse as ov
|
| 28 |
+
print(f'omicverse version:{ov.__version__}')
|
| 29 |
+
import scanpy as sc
|
| 30 |
+
print(f'scanpy version:{sc.__version__}')
|
| 31 |
+
ov.ov_plot_set()
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# ## Loading data
|
| 35 |
+
#
|
| 36 |
+
# The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics ([here](http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz) from this [webpage](https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k)). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
|
| 37 |
+
#
|
| 38 |
+
|
| 39 |
+
# In[2]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
# !mkdir data
|
| 43 |
+
# !wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 44 |
+
# !cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 45 |
+
# !mkdir write
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# Read in the count matrix into an AnnData object, which holds many slots for annotations and different representations of the data. It also comes with its own HDF5-based file format: `.h5ad`.
|
| 49 |
+
|
| 50 |
+
# In[3]:
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
adata = sc.read_10x_mtx(
|
| 54 |
+
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
|
| 55 |
+
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
|
| 56 |
+
cache=True) # write a cache file for faster subsequent reading
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
# ## Data preprocessing
|
| 60 |
+
#
|
| 61 |
+
# Here, we use `ov.single.scanpy_lazy` to preprocess the raw data of scRNA-seq, it included filter the doublets cells, normalizing counts per cell, log1p, extracting highly variable genes, and cluster of cells calculation.
|
| 62 |
+
#
|
| 63 |
+
# But if you want to experience step-by-step preprocessing, we also provide more detailed preprocessing steps here, please refer to our [preprocess chapter](https://omicverse.readthedocs.io/en/latest/Tutorials-single/t_preprocess/) for a detailed explanation.
|
| 64 |
+
#
|
| 65 |
+
# We stored the raw counts in `count` layers, and the raw data in `adata.raw.to_adata()`.
|
| 66 |
+
|
| 67 |
+
# In[4]:
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
#adata=ov.single.scanpy_lazy(adata)
|
| 71 |
+
|
| 72 |
+
#quantity control
|
| 73 |
+
adata=ov.pp.qc(adata,
|
| 74 |
+
tresh={'mito_perc': 0.05, 'nUMIs': 500, 'detected_genes': 250})
|
| 75 |
+
#normalize and high variable genes (HVGs) calculated
|
| 76 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 77 |
+
|
| 78 |
+
#save the whole genes and filter the non-HVGs
|
| 79 |
+
adata.raw = adata
|
| 80 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 81 |
+
|
| 82 |
+
#scale the adata.X
|
| 83 |
+
ov.pp.scale(adata)
|
| 84 |
+
|
| 85 |
+
#Dimensionality Reduction
|
| 86 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 87 |
+
|
| 88 |
+
#Neighbourhood graph construction
|
| 89 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 90 |
+
use_rep='scaled|original|X_pca')
|
| 91 |
+
|
| 92 |
+
#clusters
|
| 93 |
+
sc.tl.leiden(adata)
|
| 94 |
+
|
| 95 |
+
#Dimensionality Reduction for visualization(X_mde=X_umap+GPU)
|
| 96 |
+
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 97 |
+
adata
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# ## Cell annotate automatically
|
| 101 |
+
#
|
| 102 |
+
# We create a pySCSA object from the `adata`, and we need to set some parameter to annotate correctly.
|
| 103 |
+
#
|
| 104 |
+
# In normal annotate, we set `celltype`=`'normal'` and `target`=`'cellmarker'` or `'panglaodb'` to perform the cell annotate.
|
| 105 |
+
#
|
| 106 |
+
# But in cancer annotate, we need to set the `celltype`=`'cancer'` and `target`=`'cancersea'` to perform the cell annotate.
|
| 107 |
+
#
|
| 108 |
+
# <div class="admonition note">
|
| 109 |
+
# <p class="admonition-title">Note</p>
|
| 110 |
+
# <p>
|
| 111 |
+
# The annotation with SCSA need to download the database at first. It can be downloaded automatically. But sometimes you will have problems with network errors.
|
| 112 |
+
# </p>
|
| 113 |
+
# </div>
|
| 114 |
+
#
|
| 115 |
+
# The database can be downloaded from [figshare](https://figshare.com/ndownloader/files/41369037) or [Google Drive](https://drive.google.com/drive/folders/1pqyuCp8mTXDFRGUkX8iDdPAg45JHvheF?usp=sharing). And you need to set parameter `model_path`=`'path'`
|
| 116 |
+
|
| 117 |
+
# In[5]:
|
| 118 |
+
|
| 119 |
+
|
| 120 |
+
scsa=ov.single.pySCSA(adata=adata,
|
| 121 |
+
foldchange=1.5,
|
| 122 |
+
pvalue=0.01,
|
| 123 |
+
celltype='normal',
|
| 124 |
+
target='cellmarker',
|
| 125 |
+
tissue='All',
|
| 126 |
+
model_path='temp/pySCSA_2023_v2_plus.db'
|
| 127 |
+
)
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# In the previous cell clustering we used the leiden algorithm, so here we specify that the type is set to leiden. if you are using louvain, please change it. And, we will annotate all clusters, if you only want to annotate a few of the classes, please follow `'[1]'`, `'[1,2,3]'`, `'[...]'` Enter in the format.
|
| 131 |
+
#
|
| 132 |
+
# `rank_rep` means the `sc.tl.rank_genes_groups(adata, clustertype, method='wilcoxon')`, if we provided the `rank_genes_groups` in adata.uns, `rank_rep` can be set as `False`
|
| 133 |
+
|
| 134 |
+
# In[6]:
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
anno=scsa.cell_anno(clustertype='leiden',
|
| 138 |
+
cluster='all',rank_rep=True)
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
# We can query only the better annotated results
|
| 142 |
+
|
| 143 |
+
# In[7]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
scsa.cell_auto_anno(adata,key='scsa_celltype_cellmarker')
|
| 147 |
+
|
| 148 |
+
|
| 149 |
+
# We can also use `panglaodb` as target to annotate the celltype
|
| 150 |
+
|
| 151 |
+
# In[8]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
scsa=ov.single.pySCSA(adata=adata,
|
| 155 |
+
foldchange=1.5,
|
| 156 |
+
pvalue=0.01,
|
| 157 |
+
celltype='normal',
|
| 158 |
+
target='panglaodb',
|
| 159 |
+
tissue='All',
|
| 160 |
+
model_path='temp/pySCSA_2023_v2_plus.db'
|
| 161 |
+
|
| 162 |
+
)
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
# In[9]:
|
| 166 |
+
|
| 167 |
+
|
| 168 |
+
res=scsa.cell_anno(clustertype='leiden',
|
| 169 |
+
cluster='all',rank_rep=True)
|
| 170 |
+
|
| 171 |
+
|
| 172 |
+
# We can query only the better annotated results
|
| 173 |
+
|
| 174 |
+
# In[10]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
scsa.cell_anno_print()
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
# In[11]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
scsa.cell_auto_anno(adata,key='scsa_celltype_panglaodb')
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
# Here, we introduce the dimensionality reduction visualisation function `ov.utils.embedding`, which is similar to `scanpy.pl.embedding`, except that when we set `frameon='small'`, we scale the axes to the bottom-left corner and scale the colourbar to the bottom-right corner.
|
| 187 |
+
#
|
| 188 |
+
# - adata: the anndata object
|
| 189 |
+
# - basis: the visualized embedding stored in adata.obsm
|
| 190 |
+
# - color: the visualized obs/var
|
| 191 |
+
# - legend_loc: the location of legend, if you set None, it will be visualized in right.
|
| 192 |
+
# - frameon: it can be set `small`, False or None
|
| 193 |
+
# - legend_fontoutline: the outline in the text of legend.
|
| 194 |
+
# - palette: Different categories of colours, we have a number of different colours preset in omicverse, including `ov.utils.palette()`, `ov.utils.red_color`, `ov.utils.blue_color`, `ov.utils.green_color`, `ov. utils.orange_color`. The preset colours can help you achieve a more beautiful visualisation.
|
| 195 |
+
|
| 196 |
+
# In[12]:
|
| 197 |
+
|
| 198 |
+
|
| 199 |
+
ov.utils.embedding(adata,
|
| 200 |
+
basis='X_mde',
|
| 201 |
+
color=['leiden','scsa_celltype_cellmarker','scsa_celltype_panglaodb'],
|
| 202 |
+
legend_loc='on data',
|
| 203 |
+
frameon='small',
|
| 204 |
+
legend_fontoutline=2,
|
| 205 |
+
palette=ov.utils.palette()[14:],
|
| 206 |
+
)
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
# If you want to draw stacked histograms of cell type proportions, you first need to colour the groups you intend to draw using `ov.utils.embedding`. Then use `ov.utils.plot_cellproportion` to specify the groups you want to plot, and you can see a plot of cell proportions in the different groups
|
| 210 |
+
|
| 211 |
+
# In[13]:
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
#Randomly designate the first 1000 cells as group B and the rest as group A
|
| 215 |
+
adata.obs['group']='A'
|
| 216 |
+
adata.obs.loc[adata.obs.index[:1000],'group']='B'
|
| 217 |
+
#Colored
|
| 218 |
+
ov.utils.embedding(adata,
|
| 219 |
+
basis='X_mde',
|
| 220 |
+
color=['group'],
|
| 221 |
+
frameon='small',legend_fontoutline=2,
|
| 222 |
+
palette=ov.utils.red_color,
|
| 223 |
+
)
|
| 224 |
+
|
| 225 |
+
|
| 226 |
+
# In[14]:
|
| 227 |
+
|
| 228 |
+
|
| 229 |
+
ov.utils.plot_cellproportion(adata=adata,celltype_clusters='scsa_celltype_cellmarker',
|
| 230 |
+
visual_clusters='group',
|
| 231 |
+
visual_name='group',figsize=(2,4))
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
# Of course, we also provide another downscaled visualisation of the graph using `ov.utils.plot_embedding_celltype`
|
| 235 |
+
|
| 236 |
+
# In[15]:
|
| 237 |
+
|
| 238 |
+
|
| 239 |
+
ov.utils.plot_embedding_celltype(adata,figsize=None,basis='X_mde',
|
| 240 |
+
celltype_key='scsa_celltype_cellmarker',
|
| 241 |
+
title=' Cell type',
|
| 242 |
+
celltype_range=(2,6),
|
| 243 |
+
embedding_range=(4,10),)
|
| 244 |
+
|
| 245 |
+
|
| 246 |
+
# We calculated the ratio of observed to expected cell numbers (Ro/e) for each cluster in different tissues to quantify the tissue preference of each cluster (Guo et al., 2018; Zhang et al., 2018). The expected cell num- bers for each combination of cell clusters and tissues were obtained from the chi-square test. One cluster was identified as being enriched in a specific tissue if Ro/e>1.
|
| 247 |
+
#
|
| 248 |
+
# The Ro/e function was wrote by `Haihao Zhang`.
|
| 249 |
+
|
| 250 |
+
# In[16]:
|
| 251 |
+
|
| 252 |
+
|
| 253 |
+
roe=ov.utils.roe(adata,sample_key='group',cell_type_key='scsa_celltype_cellmarker')
|
| 254 |
+
|
| 255 |
+
|
| 256 |
+
# In[40]:
|
| 257 |
+
|
| 258 |
+
|
| 259 |
+
import seaborn as sns
|
| 260 |
+
import matplotlib.pyplot as plt
|
| 261 |
+
fig, ax = plt.subplots(figsize=(2,4))
|
| 262 |
+
|
| 263 |
+
transformed_roe = roe.copy()
|
| 264 |
+
transformed_roe = transformed_roe.applymap(
|
| 265 |
+
lambda x: '+++' if x >= 2 else ('++' if x >= 1.5 else ('+' if x >= 1 else '+/-')))
|
| 266 |
+
|
| 267 |
+
sns.heatmap(roe, annot=transformed_roe, cmap='RdBu_r', fmt='',
|
| 268 |
+
cbar=True, ax=ax,vmin=0.5,vmax=1.5,cbar_kws={'shrink':0.5})
|
| 269 |
+
plt.xticks(fontsize=12)
|
| 270 |
+
plt.yticks(fontsize=12)
|
| 271 |
+
|
| 272 |
+
plt.xlabel('Group',fontsize=13)
|
| 273 |
+
plt.ylabel('Cell type',fontsize=13)
|
| 274 |
+
plt.title('Ro/e',fontsize=13)
|
| 275 |
+
|
| 276 |
+
|
| 277 |
+
# ## Cell annotate manually
|
| 278 |
+
#
|
| 279 |
+
# In order to compare the accuracy of our automatic annotations, we will here use marker genes to manually annotate the cluster and compare the accuracy of the pySCSA and manual.
|
| 280 |
+
#
|
| 281 |
+
# We need to prepare a marker's dict at first
|
| 282 |
+
|
| 283 |
+
# In[38]:
|
| 284 |
+
|
| 285 |
+
|
| 286 |
+
res_marker_dict={
|
| 287 |
+
'Megakaryocyte':['ITGA2B','ITGB3'],
|
| 288 |
+
'Dendritic cell':['CLEC10A','IDO1'],
|
| 289 |
+
'Monocyte' :['S100A8','S100A9','LST1',],
|
| 290 |
+
'Macrophage':['CSF1R','CD68'],
|
| 291 |
+
'B cell':['MS4A1','CD79A','MZB1',],
|
| 292 |
+
'NK/NKT cell':['GNLY','KLRD1'],
|
| 293 |
+
'CD8+T cell':['CD8A','CD8B'],
|
| 294 |
+
'Treg':['CD4','CD40LG','IL7R','FOXP3','IL2RA'],
|
| 295 |
+
'CD4+T cell':['PTPRC','CD3D','CD3E'],
|
| 296 |
+
|
| 297 |
+
}
|
| 298 |
+
|
| 299 |
+
|
| 300 |
+
# We then calculated the expression of marker genes in each cluster and the fraction
|
| 301 |
+
|
| 302 |
+
# In[39]:
|
| 303 |
+
|
| 304 |
+
|
| 305 |
+
sc.tl.dendrogram(adata,'leiden')
|
| 306 |
+
sc.pl.dotplot(adata, res_marker_dict, 'leiden',
|
| 307 |
+
dendrogram=True,standard_scale='var')
|
| 308 |
+
|
| 309 |
+
|
| 310 |
+
# Based on the dotplot, we name each cluster according `ov.single.scanpy_cellanno_from_dict`
|
| 311 |
+
|
| 312 |
+
# In[40]:
|
| 313 |
+
|
| 314 |
+
|
| 315 |
+
# create a dictionary to map cluster to annotation label
|
| 316 |
+
cluster2annotation = {
|
| 317 |
+
'0': 'T cell',
|
| 318 |
+
'1': 'T cell',
|
| 319 |
+
'2': 'Monocyte',#Germ-cell(Oid)
|
| 320 |
+
'3': 'B cell',#Germ-cell(Oid)
|
| 321 |
+
'4': 'T cell',
|
| 322 |
+
'5': 'Macrophage',
|
| 323 |
+
'6': 'NKT cells',
|
| 324 |
+
'7': 'T cell',
|
| 325 |
+
'8':'Monocyte',
|
| 326 |
+
'9':'Dendritic cell',
|
| 327 |
+
'10':'Megakaryocyte',
|
| 328 |
+
|
| 329 |
+
}
|
| 330 |
+
ov.single.scanpy_cellanno_from_dict(adata,anno_dict=cluster2annotation,
|
| 331 |
+
clustertype='leiden')
|
| 332 |
+
|
| 333 |
+
|
| 334 |
+
# ## Compare the pySCSA and Manual
|
| 335 |
+
#
|
| 336 |
+
# We can see that the auto-annotation results are almost identical to the manual annotation, the only difference is between monocyte and macrophages, but in the previous auto-annotation results, pySCSA gives the option of `monocyte|macrophage`, so it can be assumed that pySCSA performs better on the pbmc3k data
|
| 337 |
+
|
| 338 |
+
# In[52]:
|
| 339 |
+
|
| 340 |
+
|
| 341 |
+
ov.utils.embedding(adata,
|
| 342 |
+
basis='X_mde',
|
| 343 |
+
color=['major_celltype','scsa_celltype_cellmarker'],
|
| 344 |
+
legend_loc='on data', frameon='small',legend_fontoutline=2,
|
| 345 |
+
palette=ov.utils.palette()[14:],
|
| 346 |
+
)
|
| 347 |
+
|
| 348 |
+
|
| 349 |
+
# We can use `get_celltype_marker` to obtain the marker of each celltype
|
| 350 |
+
|
| 351 |
+
# In[42]:
|
| 352 |
+
|
| 353 |
+
|
| 354 |
+
marker_dict=ov.single.get_celltype_marker(adata,clustertype='scsa_celltype_cellmarker')
|
| 355 |
+
marker_dict.keys()
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
# In[43]:
|
| 359 |
+
|
| 360 |
+
|
| 361 |
+
marker_dict['B cell']
|
| 362 |
+
|
| 363 |
+
|
| 364 |
+
# ## The tissue name in database
|
| 365 |
+
#
|
| 366 |
+
# For annotation of cell types in specific tissues, we can query the tissues available in the database using `get_model_tissue`.
|
| 367 |
+
|
| 368 |
+
# In[44]:
|
| 369 |
+
|
| 370 |
+
|
| 371 |
+
scsa.get_model_tissue()
|
| 372 |
+
|
| 373 |
+
|
| 374 |
+
# In[ ]:
|
| 375 |
+
|
| 376 |
+
|
| 377 |
+
|
| 378 |
+
|
ovrawm/t_cellfate.txt
ADDED
|
@@ -0,0 +1,218 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Identify the driver regulators of cell fate decisions
|
| 5 |
+
# CEFCON is a computational tool for deciphering driver regulators of cell fate decisions from single-cell RNA-seq data. It takes a prior gene interaction network and expression profiles from scRNA-seq data associated with a given developmental trajectory as inputs, and consists of three main components, including cell-lineage-specific gene regulatory network (GRN) construction, driver regulator identification and regulon-like gene module (RGM) identification.
|
| 6 |
+
#
|
| 7 |
+
# Check out [(Wang et al., Nature Communications, 2023)](https://www.nature.com/articles/s41467-023-44103-3) for the detailed methods and applications.
|
| 8 |
+
#
|
| 9 |
+
# Code: [https://github.com/WPZgithub/CEFCON](https://github.com/WPZgithub/CEFCON)
|
| 10 |
+
#
|
| 11 |
+
|
| 12 |
+
# In[1]:
|
| 13 |
+
|
| 14 |
+
|
| 15 |
+
import omicverse as ov
|
| 16 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 17 |
+
import scanpy as sc
|
| 18 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 19 |
+
import pandas as pd
|
| 20 |
+
from tqdm.auto import tqdm
|
| 21 |
+
ov.plot_set()
|
| 22 |
+
|
| 23 |
+
|
| 24 |
+
# # Data loading and processing
|
| 25 |
+
# Here, we use the mouse hematopoiesis data provided by [Nestorowa et al. (2016, Blood).](https://doi.org/10.1182/blood-2016-05-716480)
|
| 26 |
+
#
|
| 27 |
+
# **The scRNA-seq data requires processing to extract lineage information for the CEFCON analysis.** Please refer to the [original notebook](https://github.com/WPZgithub/CEFCON/blob/e74d2d248b88fb3349023d1a97d3cc8a52cc4060/notebooks/preprocessing_nestorowa16_data.ipynb) for detailed instructions on preprocessing scRNA-seq data.
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
adata = ov.single.mouse_hsc_nestorowa16()
|
| 33 |
+
adata
|
| 34 |
+
|
| 35 |
+
|
| 36 |
+
# CEFCON fully exploit an available global and **context-free gene interaction network** as prior knowledge, from which we extract the cell-lineage-specific gene interactions according to the gene expression profiles derived from scRNA-seq data associated with a given developmental trajectory.
|
| 37 |
+
#
|
| 38 |
+
# You can download the prior network in the [zenodo](https://zenodo.org/records/8013900). **CEFCON only provides the prior network for human and mosue data anaylsis**. For other species, you should provide the prior network mannully.
|
| 39 |
+
#
|
| 40 |
+
# The author of CEFCON has provided several prior networks here; however, 'nichenet' yields the best results.
|
| 41 |
+
|
| 42 |
+
# In[3]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
prior_network = ov.single.load_human_prior_interaction_network(dataset='nichenet')
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# **In the scRNA-seq analysis of human data, you should not run this step. Running it may change the gene symbol and result in errors.**
|
| 49 |
+
#
|
| 50 |
+
#
|
| 51 |
+
#
|
| 52 |
+
#
|
| 53 |
+
|
| 54 |
+
# In[4]:
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
# Convert the gene symbols of the prior gene interaction network to the mouse gene symbols
|
| 58 |
+
prior_network = ov.single.convert_human_to_mouse_network(prior_network,server_name='asia')
|
| 59 |
+
prior_network
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
# In[12]:
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
prior_network.to_csv('result/combined_network_Mouse.txt.gz',sep='\t')
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
# Alternatively, you can directly specify the file path of the input prior interaction network and import the specified file.
|
| 69 |
+
|
| 70 |
+
# In[3]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
#prior_network = './Reference_Networks/combined_network_Mouse.txt'
|
| 74 |
+
prior_network=ov.read('result/combined_network_Mouse.txt.gz',index_col=0)
|
| 75 |
+
|
| 76 |
+
|
| 77 |
+
# # Training CEFCON model
|
| 78 |
+
#
|
| 79 |
+
# We recommend using GRUOBI to solve the integer linear programming (ILP) problem when identifying driver genes. GUROBI is a commercial solver that requires licenses to run. Thankfully, it provides free licenses in academia, as well as trial licenses outside academia. If there is no problem about the licenses, you need to install the `gurobipy` package.
|
| 80 |
+
#
|
| 81 |
+
# If difficulties arise while using GUROBI, the non-commercial solver, SCIP, will be employed as an alternative. But the use of SCIP does not come with a guarantee of achieving a successful solutio
|
| 82 |
+
#
|
| 83 |
+
# **By default, the program will verify the availability of GRUOBI. If GRUOBI is not accessible, it will automatically switch the solver to SCIP.**
|
| 84 |
+
#
|
| 85 |
+
|
| 86 |
+
# In[4]:
|
| 87 |
+
|
| 88 |
+
|
| 89 |
+
CEFCON_obj = ov.single.pyCEFCON(adata, prior_network, repeats=5, solver='GUROBI')
|
| 90 |
+
CEFCON_obj
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
# Construct cell-lineage-specific GRNs
|
| 94 |
+
|
| 95 |
+
# In[5]:
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
CEFCON_obj.preprocess()
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
# Lineage-by-lineage computation:
|
| 102 |
+
|
| 103 |
+
# In[6]:
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
CEFCON_obj.train()
|
| 107 |
+
|
| 108 |
+
|
| 109 |
+
# In[9]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
# Idenytify driver regulators for each lineage
|
| 113 |
+
CEFCON_obj.predicted_driver_regulators()
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
# We can find out the driver regulators identified by CEFCON.
|
| 117 |
+
|
| 118 |
+
# In[10]:
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
CEFCON_obj.cefcon_results_dict['E_pseudotime'].driver_regulator.head()
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# In[11]:
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
CEFCON_obj.predicted_RGM()
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# # Downstream analysis
|
| 131 |
+
|
| 132 |
+
# In[12]:
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
CEFCON_obj.cefcon_results_dict['E_pseudotime']
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
# In[13]:
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
lineage = 'E_pseudotime'
|
| 142 |
+
result = CEFCON_obj.cefcon_results_dict[lineage]
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
# Plot gene embedding clusters
|
| 146 |
+
|
| 147 |
+
# In[20]:
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
gene_ad=sc.AnnData(result.gene_embedding)
|
| 151 |
+
sc.pp.neighbors(gene_ad, n_neighbors=30, use_rep='X')
|
| 152 |
+
# Higher resolutions lead to more communities, while lower resolutions lead to fewer communities.
|
| 153 |
+
sc.tl.leiden(gene_ad, resolution=1)
|
| 154 |
+
sc.tl.umap(gene_ad, n_components=2, min_dist=0.3)
|
| 155 |
+
|
| 156 |
+
|
| 157 |
+
# In[27]:
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
ov.utils.embedding(gene_ad,basis='X_umap',legend_loc='on data',
|
| 161 |
+
legend_fontsize=8, legend_fontoutline=2,
|
| 162 |
+
color='leiden',frameon='small',title='Leiden clustering using CEFCON\nderived gene embeddings')
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
# Plot influence scores of driver regulators
|
| 166 |
+
|
| 167 |
+
# In[40]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
import matplotlib.pyplot as plt
|
| 171 |
+
import seaborn as sns
|
| 172 |
+
data_for_plot = result.driver_regulator[result.driver_regulator['is_driver_regulator']]
|
| 173 |
+
data_for_plot = data_for_plot[0:20]
|
| 174 |
+
|
| 175 |
+
plt.figure(figsize=(2, 20 * 0.2))
|
| 176 |
+
sns.set_theme(style='ticks', font_scale=0.5)
|
| 177 |
+
|
| 178 |
+
ax = sns.barplot(x='influence_score', y=data_for_plot.index, data=data_for_plot, orient='h',
|
| 179 |
+
palette=sns.color_palette(f"ch:start=.5,rot=-.5,reverse=1,dark=0.4", n_colors=20))
|
| 180 |
+
ax.set_title(result.name)
|
| 181 |
+
ax.set_xlabel('Influence score')
|
| 182 |
+
ax.set_ylabel('Driver regulators')
|
| 183 |
+
|
| 184 |
+
ax.spines['left'].set_position(('outward', 10))
|
| 185 |
+
ax.spines['bottom'].set_position(('outward', 10))
|
| 186 |
+
plt.xticks(fontsize=12)
|
| 187 |
+
plt.yticks(fontsize=12)
|
| 188 |
+
|
| 189 |
+
plt.grid(False)
|
| 190 |
+
#设置spines可视化情况
|
| 191 |
+
ax.spines['top'].set_visible(False)
|
| 192 |
+
ax.spines['right'].set_visible(False)
|
| 193 |
+
ax.spines['bottom'].set_visible(True)
|
| 194 |
+
ax.spines['left'].set_visible(True)
|
| 195 |
+
|
| 196 |
+
plt.title('E_pseudotime',fontsize=12)
|
| 197 |
+
plt.xlabel('Influence score',fontsize=12)
|
| 198 |
+
plt.ylabel('Driver regulon',fontsize=12)
|
| 199 |
+
|
| 200 |
+
sns.despine()
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
# In[41]:
|
| 204 |
+
|
| 205 |
+
|
| 206 |
+
result.plot_driver_genes_Venn()
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
# Plot heat map of the activity matrix of RGMs
|
| 210 |
+
|
| 211 |
+
# In[42]:
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
adata_lineage = adata[adata.obs_names[adata.obs[result.name].notna()],:]
|
| 215 |
+
|
| 216 |
+
result.plot_RGM_activity_heatmap(cell_label=adata_lineage.obs['cell_type_finely'],
|
| 217 |
+
type='out',col_cluster=True,bbox_to_anchor=(1.48, 0.25))
|
| 218 |
+
|
ovrawm/t_cellfate_gene.txt
ADDED
|
@@ -0,0 +1,468 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Timing-associated genes analysis with cellfategenie
|
| 5 |
+
#
|
| 6 |
+
# In our single-cell analysis, we analyse the underlying temporal state in the cell, which we call pseudotime. and identifying the genes associated with pseudotime becomes the key to unravelling models of gene dynamic regulation. In traditional analysis, we would use correlation coefficients, or gene dynamics model fitting. The correlation coefficient approach will have a preference for genes at the beginning and end of the time series, and the gene dynamics model requires RNA velocity information. Unbiased identification of chronosequence-related genes, as well as the need for no additional dependency information, has become a challenge in current chronosequence analyses.
|
| 7 |
+
#
|
| 8 |
+
# Here, we developed CellFateGenie, which first removes potential noise from the data through metacells, and then constructs an adaptive ridge regression model to find the minimum set of genes needed to satisfy the timing fit.CellFateGenie has similar accuracy to gene dynamics models while eliminating preferences for the start and end of the time series.
|
| 9 |
+
#
|
| 10 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1Q1Sk5lGCBGBWS5Bs2kncAq9ZbjaDzSR4?usp=sharing
|
| 11 |
+
|
| 12 |
+
# In[1]:
|
| 13 |
+
|
| 14 |
+
|
| 15 |
+
import omicverse as ov
|
| 16 |
+
import scvelo as scv
|
| 17 |
+
import matplotlib.pyplot as plt
|
| 18 |
+
ov.ov_plot_set()
|
| 19 |
+
|
| 20 |
+
|
| 21 |
+
# ## Data preprocessed
|
| 22 |
+
#
|
| 23 |
+
# We using dataset of dentategyrus in scvelo to demonstrate the timing-associated genes analysis. Firstly, We use `ov.pp.qc` and `ov.pp.preprocess` to preprocess the dataset.
|
| 24 |
+
#
|
| 25 |
+
# Then we use `ov.pp.scale` and `ov.pp.pca` to analysis the principal component of the data
|
| 26 |
+
|
| 27 |
+
# In[18]:
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
adata = scv.datasets.dentategyrus()
|
| 31 |
+
adata
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# In[19]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
adata=ov.pp.qc(adata,
|
| 38 |
+
tresh={'mito_perc': 0.15, 'nUMIs': 500, 'detected_genes': 250},
|
| 39 |
+
)
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
# In[20]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
ov.utils.store_layers(adata,layers='counts')
|
| 46 |
+
adata
|
| 47 |
+
|
| 48 |
+
|
| 49 |
+
# In[21]:
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',
|
| 53 |
+
n_HVGs=2000)
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
# In[22]:
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
adata.raw = adata
|
| 60 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 61 |
+
adata
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# In[23]:
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
ov.pp.scale(adata)
|
| 68 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 69 |
+
|
| 70 |
+
adata.obsm["X_mde_pca"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# In[24]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
adata=adata.raw.to_adata()
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# In[25]:
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
fig, ax = plt.subplots(figsize=(3,3))
|
| 83 |
+
ov.utils.embedding(adata,
|
| 84 |
+
basis='X_mde_pca',frameon='small',
|
| 85 |
+
color=['clusters'],show=False,ax=ax)
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
# ## Meta-cells calculated
|
| 89 |
+
#
|
| 90 |
+
# To reduce the noisy of the raw dataset and improve the accuracy of the regrssion model. We using `SEACells` to perform the Meta-cells calculated.
|
| 91 |
+
|
| 92 |
+
# In[451]:
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
import SEACells
|
| 96 |
+
adata=adata[adata.obs['clusters']!='Endothelial']
|
| 97 |
+
model = SEACells.core.SEACells(adata,
|
| 98 |
+
build_kernel_on='scaled|original|X_pca',
|
| 99 |
+
n_SEACells=200,
|
| 100 |
+
n_waypoint_eigs=10,
|
| 101 |
+
convergence_epsilon = 1e-5)
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
# In[452]:
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
model.construct_kernel_matrix()
|
| 108 |
+
M = model.kernel_matrix
|
| 109 |
+
# Initialize archetypes
|
| 110 |
+
model.initialize_archetypes()
|
| 111 |
+
|
| 112 |
+
|
| 113 |
+
# In[453]:
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
model.fit(min_iter=10, max_iter=50)
|
| 117 |
+
|
| 118 |
+
|
| 119 |
+
# The model will stop early, we can use `model.step` to force the model run additional iterations. Usually, 100 iters can get the best metacells.
|
| 120 |
+
|
| 121 |
+
# In[454]:
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# Check for convergence
|
| 125 |
+
get_ipython().run_line_magic('matplotlib', 'inline')
|
| 126 |
+
model.plot_convergence()
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
# In[489]:
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# You can force the model to run additional iterations step-wise using the .step() function
|
| 133 |
+
print(f'Run for {len(model.RSS_iters)} iterations')
|
| 134 |
+
for _ in range(10):
|
| 135 |
+
model.step()
|
| 136 |
+
print(f'Run for {len(model.RSS_iters)} iterations')
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
# In[490]:
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
# Check for convergence
|
| 143 |
+
get_ipython().run_line_magic('matplotlib', 'inline')
|
| 144 |
+
model.plot_convergence()
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
# In[491]:
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
get_ipython().run_line_magic('matplotlib', 'inline')
|
| 151 |
+
SEACells.plot.plot_2D(adata, key='X_mde_pca', colour_metacells=False,
|
| 152 |
+
figsize=(4,4),cell_size=20,title='Dentategyrus Metacells',
|
| 153 |
+
)
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# We notice the shape of raw anndata not consistent with the HVGs anndata.
|
| 157 |
+
|
| 158 |
+
# In[492]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
adata.raw=adata.copy()
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
# And we use `SEACells.core.summarize_by_soft_SEACell` to get the normalized metacells
|
| 165 |
+
|
| 166 |
+
# In[493]:
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
SEACell_soft_ad = SEACells.core.summarize_by_soft_SEACell(adata, model.A_,
|
| 170 |
+
celltype_label='clusters',
|
| 171 |
+
summarize_layer='raw', minimum_weight=0.05)
|
| 172 |
+
SEACell_soft_ad
|
| 173 |
+
|
| 174 |
+
|
| 175 |
+
# We visualized the metacells with PCA and UMAP
|
| 176 |
+
|
| 177 |
+
# In[494]:
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
import scanpy as sc
|
| 181 |
+
SEACell_soft_ad.raw=SEACell_soft_ad.copy()
|
| 182 |
+
sc.pp.highly_variable_genes(SEACell_soft_ad, n_top_genes=2000, inplace=True)
|
| 183 |
+
SEACell_soft_ad=SEACell_soft_ad[:,SEACell_soft_ad.var.highly_variable]
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
# In[495]:
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
ov.pp.scale(SEACell_soft_ad)
|
| 190 |
+
ov.pp.pca(SEACell_soft_ad,layer='scaled',n_pcs=50)
|
| 191 |
+
sc.pp.neighbors(SEACell_soft_ad, use_rep='scaled|original|X_pca')
|
| 192 |
+
sc.tl.umap(SEACell_soft_ad)
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
# And we can use the raw color of anndata.
|
| 196 |
+
|
| 197 |
+
# In[496]:
|
| 198 |
+
|
| 199 |
+
|
| 200 |
+
SEACell_soft_ad.obs['celltype']=SEACell_soft_ad.obs['celltype'].astype('category')
|
| 201 |
+
SEACell_soft_ad.obs['celltype']=SEACell_soft_ad.obs['celltype'].cat.reorder_categories(adata.obs['clusters'].cat.categories)
|
| 202 |
+
SEACell_soft_ad.uns['celltype_colors']=adata.uns['clusters_colors']
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
# In[15]:
|
| 206 |
+
|
| 207 |
+
|
| 208 |
+
import matplotlib.pyplot as plt
|
| 209 |
+
fig, ax = plt.subplots(figsize=(3,3))
|
| 210 |
+
ov.utils.embedding(SEACell_soft_ad,
|
| 211 |
+
basis='X_umap',
|
| 212 |
+
color=["celltype"],
|
| 213 |
+
title='Meta Celltype',
|
| 214 |
+
frameon='small',
|
| 215 |
+
legend_fontsize=12,
|
| 216 |
+
#palette=ov.utils.palette()[11:],
|
| 217 |
+
ax=ax,
|
| 218 |
+
show=False)
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
# ## Pseudotime calculated
|
| 222 |
+
#
|
| 223 |
+
# Accurately calculating the pseudotime in metacells is another challenge we need to face, here we use pyVIA to complete the calculation of the pseudotime. Since the metacell has only 200 cells, we may not get proper proposed time series results by using the default parameters of pyVIA, so we manually adjust the relevant parameters.
|
| 224 |
+
#
|
| 225 |
+
# We need to set `jac_std_global`, `too_big_factor` and `knn` manually. If you know the origin cells, set the `root_user` is helpful too.
|
| 226 |
+
|
| 227 |
+
# In[ ]:
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
v0 = ov.single.pyVIA(adata=SEACell_soft_ad,adata_key='scaled|original|X_pca',
|
| 231 |
+
adata_ncomps=50, basis='X_umap',
|
| 232 |
+
clusters='celltype',knn=10, root_user=['nIPC','Neuroblast'],
|
| 233 |
+
dataset='group',
|
| 234 |
+
random_seed=112,is_coarse=True,
|
| 235 |
+
preserve_disconnected=True,
|
| 236 |
+
piegraph_arrow_head_width=0.05,piegraph_edgeweight_scalingfactor=2.5,
|
| 237 |
+
gene_matrix=SEACell_soft_ad.X,velo_weight=0.5,
|
| 238 |
+
edgebundle_pruning_twice=False, edgebundle_pruning=0.15,
|
| 239 |
+
jac_std_global=0.05,too_big_factor=0.05,
|
| 240 |
+
cluster_graph_pruning_std=1,
|
| 241 |
+
time_series=False,
|
| 242 |
+
)
|
| 243 |
+
|
| 244 |
+
v0.run()
|
| 245 |
+
|
| 246 |
+
|
| 247 |
+
# In[500]:
|
| 248 |
+
|
| 249 |
+
|
| 250 |
+
v0.get_pseudotime(SEACell_soft_ad)
|
| 251 |
+
|
| 252 |
+
|
| 253 |
+
# In[17]:
|
| 254 |
+
|
| 255 |
+
|
| 256 |
+
#v0.get_pseudotime(SEACell_soft_ad)
|
| 257 |
+
import matplotlib.pyplot as plt
|
| 258 |
+
fig, ax = plt.subplots(figsize=(3,3))
|
| 259 |
+
ov.utils.embedding(SEACell_soft_ad,
|
| 260 |
+
basis='X_umap',
|
| 261 |
+
color=["pt_via"],
|
| 262 |
+
title='Pseudotime',
|
| 263 |
+
frameon='small',
|
| 264 |
+
cmap='Reds',
|
| 265 |
+
#size=40,
|
| 266 |
+
legend_fontsize=12,
|
| 267 |
+
#palette=ov.utils.palette()[11:],
|
| 268 |
+
ax=ax,
|
| 269 |
+
show=False)
|
| 270 |
+
|
| 271 |
+
|
| 272 |
+
# Now we save the result of metacells for under analysis.
|
| 273 |
+
|
| 274 |
+
# In[502]:
|
| 275 |
+
|
| 276 |
+
|
| 277 |
+
SEACell_soft_ad.write_h5ad('data/tutorial_meta_den.h5ad',compression='gzip')
|
| 278 |
+
|
| 279 |
+
|
| 280 |
+
# In[2]:
|
| 281 |
+
|
| 282 |
+
|
| 283 |
+
SEACell_soft_ad=ov.utils.read('data/tutorial_meta_den.h5ad')
|
| 284 |
+
|
| 285 |
+
|
| 286 |
+
# ## Timing-associated genes analysis
|
| 287 |
+
#
|
| 288 |
+
# We have encapsulated the cellfategenie algorithm into omicverse, and we can simply use omicverse to analysis.
|
| 289 |
+
|
| 290 |
+
# In[3]:
|
| 291 |
+
|
| 292 |
+
|
| 293 |
+
cfg_obj=ov.single.cellfategenie(SEACell_soft_ad,pseudotime='pt_via')
|
| 294 |
+
cfg_obj.model_init()
|
| 295 |
+
|
| 296 |
+
|
| 297 |
+
# We used Adaptive Threshold Regression to calculate the minimum number of gene sets that would have the same accuracy as the regression model constructed for all genes.
|
| 298 |
+
|
| 299 |
+
# In[4]:
|
| 300 |
+
|
| 301 |
+
|
| 302 |
+
cfg_obj.ATR(stop=500,flux=0.01)
|
| 303 |
+
|
| 304 |
+
|
| 305 |
+
# In[5]:
|
| 306 |
+
|
| 307 |
+
|
| 308 |
+
fig,ax=cfg_obj.plot_filtering(color='#5ca8dc')
|
| 309 |
+
ax.set_title('Dentategyrus Metacells\nCellFateGenie')
|
| 310 |
+
|
| 311 |
+
|
| 312 |
+
# In[6]:
|
| 313 |
+
|
| 314 |
+
|
| 315 |
+
res=cfg_obj.model_fit()
|
| 316 |
+
|
| 317 |
+
|
| 318 |
+
# ## Visualization
|
| 319 |
+
#
|
| 320 |
+
# We prepared a series of function to visualize the result. we can use `plot_color_fitting` to observe the different cells how to transit with the pseudotime.
|
| 321 |
+
|
| 322 |
+
# In[7]:
|
| 323 |
+
|
| 324 |
+
|
| 325 |
+
cfg_obj.plot_color_fitting(type='raw',cluster_key='celltype')
|
| 326 |
+
|
| 327 |
+
|
| 328 |
+
# In[8]:
|
| 329 |
+
|
| 330 |
+
|
| 331 |
+
cfg_obj.plot_color_fitting(type='filter',cluster_key='celltype')
|
| 332 |
+
|
| 333 |
+
|
| 334 |
+
# ## Kendalltau test
|
| 335 |
+
#
|
| 336 |
+
# We can further narrow down the set of genes that satisfy the maximum regression coefficient. We used the kendalltau test to calculate the trend significance for each gene.
|
| 337 |
+
|
| 338 |
+
# In[9]:
|
| 339 |
+
|
| 340 |
+
|
| 341 |
+
kt_filter=cfg_obj.kendalltau_filter()
|
| 342 |
+
kt_filter.head()
|
| 343 |
+
|
| 344 |
+
|
| 345 |
+
# In[10]:
|
| 346 |
+
|
| 347 |
+
|
| 348 |
+
var_name=kt_filter.loc[kt_filter['pvalue']<kt_filter['pvalue'].mean()].index.tolist()
|
| 349 |
+
gt_obj=ov.single.gene_trends(SEACell_soft_ad,'pt_via',var_name)
|
| 350 |
+
gt_obj.calculate(n_convolve=10)
|
| 351 |
+
|
| 352 |
+
|
| 353 |
+
# In[11]:
|
| 354 |
+
|
| 355 |
+
|
| 356 |
+
print(f"Dimension: {len(var_name)}")
|
| 357 |
+
|
| 358 |
+
|
| 359 |
+
# In[12]:
|
| 360 |
+
|
| 361 |
+
|
| 362 |
+
fig,ax=gt_obj.plot_trend(color=ov.utils.blue_color[3])
|
| 363 |
+
ax.set_title(f'Dentategyrus meta\nCellfategenie',fontsize=13)
|
| 364 |
+
|
| 365 |
+
|
| 366 |
+
# In[14]:
|
| 367 |
+
|
| 368 |
+
|
| 369 |
+
g=ov.utils.plot_heatmap(SEACell_soft_ad,var_names=var_name,
|
| 370 |
+
sortby='pt_via',col_color='celltype',
|
| 371 |
+
n_convolve=10,figsize=(1,6),show=False)
|
| 372 |
+
|
| 373 |
+
g.fig.set_size_inches(2, 6)
|
| 374 |
+
g.fig.suptitle('CellFateGenie',x=0.25,y=0.83,
|
| 375 |
+
horizontalalignment='left',fontsize=12,fontweight='bold')
|
| 376 |
+
g.ax_heatmap.set_yticklabels(g.ax_heatmap.get_yticklabels(),fontsize=12)
|
| 377 |
+
plt.show()
|
| 378 |
+
|
| 379 |
+
|
| 380 |
+
# ## Fate Genes
|
| 381 |
+
#
|
| 382 |
+
# Unlike traditional proposed timing analyses, CellFateGenie can also access key genes/gene sets during fate transitions
|
| 383 |
+
|
| 384 |
+
# In[26]:
|
| 385 |
+
|
| 386 |
+
|
| 387 |
+
gt_obj.cal_border_cell(SEACell_soft_ad,'pt_via','celltype')
|
| 388 |
+
|
| 389 |
+
|
| 390 |
+
# In[27]:
|
| 391 |
+
|
| 392 |
+
|
| 393 |
+
bordgene_dict=gt_obj.get_multi_border_gene(SEACell_soft_ad,'celltype',
|
| 394 |
+
threshold=0.5)
|
| 395 |
+
|
| 396 |
+
|
| 397 |
+
# We use `Granule immature` and `Granule mature` to try calculated the fate related genes.
|
| 398 |
+
|
| 399 |
+
# In[30]:
|
| 400 |
+
|
| 401 |
+
|
| 402 |
+
gt_obj.get_border_gene(SEACell_soft_ad,'celltype','Granule immature','Granule mature',
|
| 403 |
+
threshold=0.5)
|
| 404 |
+
|
| 405 |
+
|
| 406 |
+
# In comparison to the `get_border_gene` function, the `get_special_border_gene` function serves the purpose of extracting exclusive fate information from two specific cell types. However, it operates with a higher degree of stringency.
|
| 407 |
+
|
| 408 |
+
# In[33]:
|
| 409 |
+
|
| 410 |
+
|
| 411 |
+
gt_obj.get_special_border_gene(SEACell_soft_ad,'celltype','Granule immature','Granule mature')
|
| 412 |
+
|
| 413 |
+
|
| 414 |
+
# We can visualize these genes.
|
| 415 |
+
|
| 416 |
+
# In[36]:
|
| 417 |
+
|
| 418 |
+
|
| 419 |
+
import matplotlib.pyplot as plt
|
| 420 |
+
g=ov.utils.plot_heatmap(SEACell_soft_ad,
|
| 421 |
+
var_names=gt_obj.get_border_gene(SEACell_soft_ad,'celltype','Granule immature','Granule mature'),
|
| 422 |
+
sortby='pt_via',col_color='celltype',yticklabels=True,
|
| 423 |
+
n_convolve=10,figsize=(1,6),show=False)
|
| 424 |
+
|
| 425 |
+
g.fig.set_size_inches(2, 4)
|
| 426 |
+
g.ax_heatmap.set_yticklabels(g.ax_heatmap.get_yticklabels(),fontsize=12)
|
| 427 |
+
plt.show()
|
| 428 |
+
|
| 429 |
+
|
| 430 |
+
# Similiarly, we can use `get_special_kernel_gene` or `get_kernel_gene` to obtain the celltype special genes.
|
| 431 |
+
|
| 432 |
+
# In[37]:
|
| 433 |
+
|
| 434 |
+
|
| 435 |
+
gt_obj.get_special_kernel_gene(SEACell_soft_ad,'celltype','Granule immature')
|
| 436 |
+
|
| 437 |
+
|
| 438 |
+
# In[42]:
|
| 439 |
+
|
| 440 |
+
|
| 441 |
+
gt_obj.get_kernel_gene(SEACell_soft_ad,
|
| 442 |
+
'celltype','Granule immature',
|
| 443 |
+
threshold=0.3,
|
| 444 |
+
num_gene=10)
|
| 445 |
+
|
| 446 |
+
|
| 447 |
+
# In[43]:
|
| 448 |
+
|
| 449 |
+
|
| 450 |
+
import matplotlib.pyplot as plt
|
| 451 |
+
g=ov.utils.plot_heatmap(SEACell_soft_ad,
|
| 452 |
+
var_names=gt_obj.get_kernel_gene(SEACell_soft_ad,
|
| 453 |
+
'celltype','Granule immature',
|
| 454 |
+
threshold=0.3,
|
| 455 |
+
num_gene=10),
|
| 456 |
+
sortby='pt_via',col_color='celltype',yticklabels=True,
|
| 457 |
+
n_convolve=10,figsize=(1,6),show=False)
|
| 458 |
+
|
| 459 |
+
g.fig.set_size_inches(2, 4)
|
| 460 |
+
g.ax_heatmap.set_yticklabels(g.ax_heatmap.get_yticklabels(),fontsize=12)
|
| 461 |
+
plt.show()
|
| 462 |
+
|
| 463 |
+
|
| 464 |
+
# In[ ]:
|
| 465 |
+
|
| 466 |
+
|
| 467 |
+
|
| 468 |
+
|
ovrawm/t_cellfate_genesets.txt
ADDED
|
@@ -0,0 +1,181 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Timing-associated geneset analysis with cellfategenie
|
| 5 |
+
#
|
| 6 |
+
# In our single-cell analysis, we analyse the underlying temporal state in the cell, which we call pseudotime. and identifying the genes associated with pseudotime becomes the key to unravelling models of gene dynamic regulation. In traditional analysis, we would use correlation coefficients, or gene dynamics model fitting. The correlation coefficient approach will have a preference for genes at the beginning and end of the time series, and the gene dynamics model requires RNA velocity information. Unbiased identification of chronosequence-related genes, as well as the need for no additional dependency information, has become a challenge in current chronosequence analyses.
|
| 7 |
+
#
|
| 8 |
+
# Here, we developed CellFateGenie, which first removes potential noise from the data through metacells, and then constructs an adaptive ridge regression model to find the minimum set of genes needed to satisfy the timing fit.CellFateGenie has similar accuracy to gene dynamics models while eliminating preferences for the start and end of the time series.
|
| 9 |
+
#
|
| 10 |
+
# We provided the AUCell to evaluate the geneset of adata
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1upcKKZHsZMS78eOliwRAddbaZ9ICXSrc?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[ ]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
import scvelo as scv
|
| 19 |
+
import matplotlib.pyplot as plt
|
| 20 |
+
ov.ov_plot_set()
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
# ## Data preprocessed
|
| 24 |
+
#
|
| 25 |
+
# We using dataset of dentategyrus in scvelo to demonstrate the timing-associated genes analysis. Firstly, We use `ov.pp.qc` and `ov.pp.preprocess` to preprocess the dataset.
|
| 26 |
+
#
|
| 27 |
+
# Then we use `ov.pp.scale` and `ov.pp.pca` to analysis the principal component of the data
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
adata=ov.read('data/tutorial_meta_den.h5ad')
|
| 33 |
+
adata=adata.raw.to_adata()
|
| 34 |
+
adata
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
# ## Genesets evaluata
|
| 38 |
+
|
| 39 |
+
# In[3]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
import omicverse as ov
|
| 43 |
+
pathway_dict=ov.utils.geneset_prepare('../placenta/genesets/GO_Biological_Process_2021.txt',organism='Mouse')
|
| 44 |
+
len(pathway_dict.keys())
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
# In[ ]:
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
##Assest all pathways
|
| 51 |
+
adata_aucs=ov.single.pathway_aucell_enrichment(adata,
|
| 52 |
+
pathways_dict=pathway_dict,
|
| 53 |
+
num_workers=8)
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
# In[11]:
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
adata_aucs.obs=adata[adata_aucs.obs.index].obs
|
| 60 |
+
adata_aucs.obsm=adata[adata_aucs.obs.index].obsm
|
| 61 |
+
adata_aucs.obsp=adata[adata_aucs.obs.index].obsp
|
| 62 |
+
adata_aucs.uns=adata[adata_aucs.obs.index].uns
|
| 63 |
+
|
| 64 |
+
adata_aucs
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# ## Timing-associated genes analysis
|
| 68 |
+
#
|
| 69 |
+
# We have encapsulated the cellfategenie algorithm into omicverse, and we can simply use omicverse to analysis.
|
| 70 |
+
|
| 71 |
+
# In[12]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
cfg_obj=ov.single.cellfategenie(adata_aucs,pseudotime='pt_via')
|
| 75 |
+
cfg_obj.model_init()
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# We used Adaptive Threshold Regression to calculate the minimum number of gene sets that would have the same accuracy as the regression model constructed for all genes.
|
| 79 |
+
|
| 80 |
+
# In[13]:
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
cfg_obj.ATR(stop=500)
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# In[14]:
|
| 87 |
+
|
| 88 |
+
|
| 89 |
+
fig,ax=cfg_obj.plot_filtering(color='#5ca8dc')
|
| 90 |
+
ax.set_title('Dentategyrus Metacells\nCellFateGenie')
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
# In[15]:
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
res=cfg_obj.model_fit()
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
# ## Visualization
|
| 100 |
+
#
|
| 101 |
+
# We prepared a series of function to visualize the result. we can use `plot_color_fitting` to observe the different cells how to transit with the pseudotime.
|
| 102 |
+
|
| 103 |
+
# In[16]:
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
cfg_obj.plot_color_fitting(type='raw',cluster_key='celltype')
|
| 107 |
+
|
| 108 |
+
|
| 109 |
+
# In[17]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
cfg_obj.plot_color_fitting(type='filter',cluster_key='celltype')
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# ## Kendalltau test
|
| 116 |
+
#
|
| 117 |
+
# We can further narrow down the set of genes that satisfy the maximum regression coefficient. We used the kendalltau test to calculate the trend significance for each gene.
|
| 118 |
+
|
| 119 |
+
# In[18]:
|
| 120 |
+
|
| 121 |
+
|
| 122 |
+
kt_filter=cfg_obj.kendalltau_filter()
|
| 123 |
+
kt_filter.head()
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
# In[21]:
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
var_name=kt_filter.loc[kt_filter['pvalue']<kt_filter['pvalue'].mean()].index.tolist()
|
| 130 |
+
gt_obj=ov.single.gene_trends(adata_aucs,'pt_via',var_name)
|
| 131 |
+
gt_obj.calculate(n_convolve=10)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# In[22]:
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
print(f"Dimension: {len(var_name)}")
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
# In[23]:
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
fig,ax=gt_obj.plot_trend(color=ov.utils.blue_color[3])
|
| 144 |
+
ax.set_title(f'Dentategyrus meta\nCellfategenie',fontsize=13)
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
# In[25]:
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
g=ov.utils.plot_heatmap(adata_aucs,var_names=var_name,
|
| 151 |
+
sortby='pt_via',col_color='celltype',
|
| 152 |
+
n_convolve=10,figsize=(1,6),show=False)
|
| 153 |
+
|
| 154 |
+
g.fig.set_size_inches(2, 6)
|
| 155 |
+
g.fig.suptitle('CellFateGenie',x=0.25,y=0.83,
|
| 156 |
+
horizontalalignment='left',fontsize=12,fontweight='bold')
|
| 157 |
+
g.ax_heatmap.set_yticklabels(g.ax_heatmap.get_yticklabels(),fontsize=12)
|
| 158 |
+
plt.show()
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
# In[32]:
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
gw_obj1=ov.utils.geneset_wordcloud(adata=adata_aucs[:,var_name],
|
| 165 |
+
cluster_key='celltype',pseudotime='pt_via',figsize=(3,6))
|
| 166 |
+
gw_obj1.get()
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
# In[33]:
|
| 170 |
+
|
| 171 |
+
|
| 172 |
+
g=gw_obj1.plot_heatmap(figwidth=6,cmap='RdBu_r')
|
| 173 |
+
plt.suptitle('CellFateGenie',x=0.18,y=0.95,
|
| 174 |
+
horizontalalignment='left',fontsize=12,fontweight='bold')
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
# In[ ]:
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
|
ovrawm/t_cellphonedb.txt
ADDED
|
@@ -0,0 +1,439 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Cell interaction with CellPhoneDB
|
| 5 |
+
#
|
| 6 |
+
# CellPhoneDB is a publicly available repository of curated receptors, ligands and their interactions in HUMAN. CellPhoneDB can be used to search for a particular ligand/receptor, or interrogate your own single-cell transcriptomics data.
|
| 7 |
+
#
|
| 8 |
+
# We made three improvements in integrating the CellPhoneDB algorithm in OmicVerse:
|
| 9 |
+
#
|
| 10 |
+
# - We have added a tutorial on analysing `anndata` based on any `anndata`.
|
| 11 |
+
# - We added prettier heatmaps, chord diagrams and network diagrams for visualising relationships between cells.
|
| 12 |
+
# - We added visualisation of ligand receptor proteins in different groups
|
| 13 |
+
#
|
| 14 |
+
# Paper: [Single-cell reconstruction of the early maternal–fetal interface in humans](https://www.nature.com/articles/s41586-018-0698-6)
|
| 15 |
+
#
|
| 16 |
+
# Code: https://github.com/ventolab/CellphoneDB
|
| 17 |
+
#
|
| 18 |
+
# This notebook will demonstrate how to use CellPhoneDB on scRNA data and visualize it.
|
| 19 |
+
|
| 20 |
+
# In[1]:
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
import scanpy as sc
|
| 24 |
+
import matplotlib.pyplot as plt
|
| 25 |
+
import pandas as pd
|
| 26 |
+
import numpy as np
|
| 27 |
+
import omicverse as ov
|
| 28 |
+
import os
|
| 29 |
+
|
| 30 |
+
ov.plot_set()
|
| 31 |
+
#print(f'cellphonedb version{cellphonedb.__version__}')
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# ## The EVT Data
|
| 35 |
+
#
|
| 36 |
+
# Th EVT data have finished the celltype annotation, it can be download from the tutorial of CellPhoneDB.
|
| 37 |
+
#
|
| 38 |
+
# Download: https://github.com/ventolab/CellphoneDB/blob/master/notebooks/data_tutorial.zip
|
| 39 |
+
#
|
| 40 |
+
|
| 41 |
+
# In[2]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
adata=sc.read('data/cpdb/normalised_log_counts.h5ad')
|
| 45 |
+
adata=adata[adata.obs['cell_labels'].isin(['eEVT','iEVT','EVT_1','EVT_2','DC','dNK1','dNK2','dNK3',
|
| 46 |
+
'VCT','VCT_CCC','VCT_fusing','VCT_p','GC','SCT'])]
|
| 47 |
+
adata
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
# In[3]:
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
ov.pl.embedding(adata,
|
| 54 |
+
basis='X_umap',
|
| 55 |
+
color='cell_labels',
|
| 56 |
+
frameon='small',
|
| 57 |
+
palette=ov.pl.red_color+ov.pl.blue_color+ov.pl.green_color+ov.pl.orange_color+ov.pl.purple_color)
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
# In[4]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
adata.X.max()
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
# We can clearly see that the maximum value of the data is a floating point number less than 10. The fact that the maximum value is not an integer means that it has been normalised, and the fact that it is less than 10 means that it has been logarithmised. Note that our data cannot be `scaled`.
|
| 67 |
+
|
| 68 |
+
# ## Export the anndata object
|
| 69 |
+
#
|
| 70 |
+
# As the input to CellPhoneDB only requires the expression matrix and cell type, we extracted only the expression matrix and cell type from adata for the next step of analysis
|
| 71 |
+
|
| 72 |
+
# In[5]:
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
sc.pp.filter_cells(adata, min_genes=200)
|
| 76 |
+
sc.pp.filter_genes(adata, min_cells=3)
|
| 77 |
+
adata1=sc.AnnData(adata.X,obs=pd.DataFrame(index=adata.obs.index),
|
| 78 |
+
var=pd.DataFrame(index=adata.var.index))
|
| 79 |
+
adata1.write_h5ad('data/cpdb/norm_log.h5ad',compression='gzip')
|
| 80 |
+
adata1
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
# ## Export the meta info of cells
|
| 84 |
+
#
|
| 85 |
+
# we construct a `DataFrame` object to export the meta info of cells. In EVT adata object, the celltypes were stored in the `obs['cell_labels']`
|
| 86 |
+
|
| 87 |
+
# In[6]:
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
#meta导出
|
| 91 |
+
df_meta = pd.DataFrame(data={'Cell':list(adata[adata1.obs.index].obs.index),
|
| 92 |
+
'cell_type':[ i for i in adata[adata1.obs.index].obs['cell_labels']]
|
| 93 |
+
})
|
| 94 |
+
df_meta.set_index('Cell', inplace=True)
|
| 95 |
+
df_meta.to_csv('data/cpdb/meta.tsv', sep = '\t')
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# ## Cell interaction analysis
|
| 99 |
+
#
|
| 100 |
+
# Now, we prepare the meta info of cells `meta.tsv` and matrix of scRNA-eq `norm_log.h5ad`, we can use the method of CellPhoneDB to calculate the interaction of each celltype in scRNA-seq data.
|
| 101 |
+
#
|
| 102 |
+
# Importantly, to avoid a series of bugs, we set the absolute path for CellPhoneDB analysis. we use `os.getcwd() ` to get the path now analysis.
|
| 103 |
+
|
| 104 |
+
# In[7]:
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
import os
|
| 108 |
+
os.getcwd()
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
# Another thing to note is that we need to download the `cellphonedb.zip` file from `cellphonedb-data` for further analysis. I have placed it in the `data/CPDB` directory, but you can place it in any path you are interested in
|
| 112 |
+
#
|
| 113 |
+
# Downloads: https://github.com/ventolab/cellphonedb-data
|
| 114 |
+
|
| 115 |
+
# In[8]:
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
cpdb_file_path = '/Users/fernandozeng/Desktop/analysis/cellphonedb-data/cellphonedb.zip'
|
| 119 |
+
meta_file_path = os.getcwd()+'/data/cpdb/meta.tsv'
|
| 120 |
+
counts_file_path = os.getcwd()+'/data/cpdb/norm_log.h5ad'
|
| 121 |
+
microenvs_file_path = None
|
| 122 |
+
active_tf_path = None
|
| 123 |
+
out_path =os.getcwd()+'/data/cpdb/test_cellphone'
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
# Now we run `cpdb_statistical_analysis_method` to predicted the cell interaction in scRNA-seq
|
| 127 |
+
|
| 128 |
+
# In[9]:
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
from cellphonedb.src.core.methods import cpdb_statistical_analysis_method
|
| 132 |
+
|
| 133 |
+
cpdb_results = cpdb_statistical_analysis_method.call(
|
| 134 |
+
cpdb_file_path = cpdb_file_path, # mandatory: CellphoneDB database zip file.
|
| 135 |
+
meta_file_path = meta_file_path, # mandatory: tsv file defining barcodes to cell label.
|
| 136 |
+
counts_file_path = counts_file_path, # mandatory: normalized count matrix - a path to the counts file, or an in-memory AnnData object
|
| 137 |
+
counts_data = 'hgnc_symbol', # defines the gene annotation in counts matrix.
|
| 138 |
+
active_tfs_file_path = active_tf_path, # optional: defines cell types and their active TFs.
|
| 139 |
+
microenvs_file_path = microenvs_file_path, # optional (default: None): defines cells per microenvironment.
|
| 140 |
+
score_interactions = True, # optional: whether to score interactions or not.
|
| 141 |
+
iterations = 1000, # denotes the number of shufflings performed in the analysis.
|
| 142 |
+
threshold = 0.1, # defines the min % of cells expressing a gene for this to be employed in the analysis.
|
| 143 |
+
threads = 10, # number of threads to use in the analysis.
|
| 144 |
+
debug_seed = 42, # debug randome seed. To disable >=0.
|
| 145 |
+
result_precision = 3, # Sets the rounding for the mean values in significan_means.
|
| 146 |
+
pvalue = 0.05, # P-value threshold to employ for significance.
|
| 147 |
+
subsampling = False, # To enable subsampling the data (geometri sketching).
|
| 148 |
+
subsampling_log = False, # (mandatory) enable subsampling log1p for non log-transformed data inputs.
|
| 149 |
+
subsampling_num_pc = 100, # Number of componets to subsample via geometric skectching (dafault: 100).
|
| 150 |
+
subsampling_num_cells = 1000, # Number of cells to subsample (integer) (default: 1/3 of the dataset).
|
| 151 |
+
separator = '|', # Sets the string to employ to separate cells in the results dataframes "cellA|CellB".
|
| 152 |
+
debug = False, # Saves all intermediate tables employed during the analysis in pkl format.
|
| 153 |
+
output_path = out_path, # Path to save results.
|
| 154 |
+
output_suffix = None # Replaces the timestamp in the output files by a user defined string in the (default: None).
|
| 155 |
+
)
|
| 156 |
+
|
| 157 |
+
|
| 158 |
+
# In[10]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
ov.utils.save(cpdb_results,'data/cpdb/gex_cpdb_test.pkl')
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
# In[5]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
cpdb_results=ov.utils.load('data/cpdb/gex_cpdb_test.pkl')
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
# ## Network of celltype analysis
|
| 171 |
+
#
|
| 172 |
+
# It is worth noting that we will be using ov for all downstream analysis, starting with cell network analysis, where we provide the `ov.single.cpdb_network_cal` function to extract interactions, and the `ov.single.cpdb_plot_network` function for very elegant visualization
|
| 173 |
+
|
| 174 |
+
# In[6]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
interaction=ov.single.cpdb_network_cal(adata = adata,
|
| 178 |
+
pvals = cpdb_results['pvalues'],
|
| 179 |
+
celltype_key = "cell_labels",)
|
| 180 |
+
|
| 181 |
+
|
| 182 |
+
# In[7]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
interaction['interaction_edges'].head()
|
| 186 |
+
|
| 187 |
+
|
| 188 |
+
# In[8]:
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
ov.plot_set()
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
# In[9]:
|
| 195 |
+
|
| 196 |
+
|
| 197 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 198 |
+
ov.pl.cpdb_heatmap(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 199 |
+
fontsize=11,
|
| 200 |
+
ax=ax,legend_kws={'fontsize':12,'bbox_to_anchor':(5, -0.9),'loc':'center left',})
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
# In[10]:
|
| 204 |
+
|
| 205 |
+
|
| 206 |
+
fig, ax = plt.subplots(figsize=(2,4))
|
| 207 |
+
ov.pl.cpdb_heatmap(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 208 |
+
source_cells=['EVT_1','EVT_2','dNK1','dNK2','dNK3'],
|
| 209 |
+
ax=ax,legend_kws={'fontsize':12,'bbox_to_anchor':(5, -0.9),'loc':'center left',})
|
| 210 |
+
|
| 211 |
+
|
| 212 |
+
# In[11]:
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
fig=ov.pl.cpdb_chord(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 216 |
+
count_min=60,fontsize=12,padding=50,radius=100,save=None,)
|
| 217 |
+
fig.show()
|
| 218 |
+
|
| 219 |
+
|
| 220 |
+
# In[12]:
|
| 221 |
+
|
| 222 |
+
|
| 223 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 224 |
+
ov.pl.cpdb_network(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 225 |
+
counts_min=60,
|
| 226 |
+
nodesize_scale=5,
|
| 227 |
+
ax=ax)
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
# In[13]:
|
| 231 |
+
|
| 232 |
+
|
| 233 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 234 |
+
ov.pl.cpdb_network(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 235 |
+
counts_min=60,
|
| 236 |
+
nodesize_scale=5,
|
| 237 |
+
source_cells=['EVT_1','EVT_2','dNK1','dNK2','dNK3'],
|
| 238 |
+
ax=ax)
|
| 239 |
+
|
| 240 |
+
|
| 241 |
+
# In[14]:
|
| 242 |
+
|
| 243 |
+
|
| 244 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 245 |
+
ov.pl.cpdb_network(adata,interaction['interaction_edges'],celltype_key='cell_labels',
|
| 246 |
+
counts_min=60,
|
| 247 |
+
nodesize_scale=5,
|
| 248 |
+
target_cells=['EVT_1','EVT_2','dNK1','dNK2','dNK3'],
|
| 249 |
+
ax=ax)
|
| 250 |
+
|
| 251 |
+
|
| 252 |
+
# In[15]:
|
| 253 |
+
|
| 254 |
+
|
| 255 |
+
ov.single.cpdb_plot_network(adata=adata,
|
| 256 |
+
interaction_edges=interaction['interaction_edges'],
|
| 257 |
+
celltype_key='cell_labels',
|
| 258 |
+
nodecolor_dict=None,title='EVT Network',
|
| 259 |
+
edgeswidth_scale=25,nodesize_scale=10,
|
| 260 |
+
pos_scale=1,pos_size=10,figsize=(6,6),
|
| 261 |
+
legend_ncol=3,legend_bbox=(0.8,0.2),legend_fontsize=10)
|
| 262 |
+
|
| 263 |
+
|
| 264 |
+
# Sometimes, the whole network you don't want to use for analysis, the sub-network is useful for analysis. we can exacted the sub-network from it.
|
| 265 |
+
#
|
| 266 |
+
# We need to exacted the sub-interaction first, we assumed that the five celltypes `['EVT_1','EVT_2','dNK1','dNK2','dNK3']` which is interested
|
| 267 |
+
|
| 268 |
+
# In[16]:
|
| 269 |
+
|
| 270 |
+
|
| 271 |
+
sub_i=interaction['interaction_edges']
|
| 272 |
+
sub_i=sub_i.loc[sub_i['SOURCE'].isin(['EVT_1','EVT_2','dNK1','dNK2','dNK3'])]
|
| 273 |
+
sub_i=sub_i.loc[sub_i['TARGET'].isin(['EVT_1','EVT_2','dNK1','dNK2','dNK3'])]
|
| 274 |
+
|
| 275 |
+
|
| 276 |
+
# Then, we exacted the sub-anndata object
|
| 277 |
+
|
| 278 |
+
# In[17]:
|
| 279 |
+
|
| 280 |
+
|
| 281 |
+
sub_adata=adata[adata.obs['cell_labels'].isin(['EVT_1','EVT_2','dNK1','dNK2','dNK3'])]
|
| 282 |
+
sub_adata
|
| 283 |
+
|
| 284 |
+
|
| 285 |
+
# Now we plot the sub-interaction network between the cells in scRNA-seq
|
| 286 |
+
|
| 287 |
+
# In[18]:
|
| 288 |
+
|
| 289 |
+
|
| 290 |
+
ov.single.cpdb_plot_network(adata=sub_adata,
|
| 291 |
+
interaction_edges=sub_i,
|
| 292 |
+
celltype_key='cell_labels',
|
| 293 |
+
nodecolor_dict=None,title='Sub-EVT Network',
|
| 294 |
+
edgeswidth_scale=25,nodesize_scale=1,
|
| 295 |
+
pos_scale=1,pos_size=10,figsize=(5,5),
|
| 296 |
+
legend_ncol=3,legend_bbox=(0.8,0.2),legend_fontsize=10)
|
| 297 |
+
|
| 298 |
+
|
| 299 |
+
# In[19]:
|
| 300 |
+
|
| 301 |
+
|
| 302 |
+
fig=ov.pl.cpdb_chord(sub_adata,sub_i,celltype_key='cell_labels',
|
| 303 |
+
count_min=10,fontsize=12,padding=60,radius=100,save=None,)
|
| 304 |
+
fig.show()
|
| 305 |
+
|
| 306 |
+
|
| 307 |
+
# In[20]:
|
| 308 |
+
|
| 309 |
+
|
| 310 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 311 |
+
ov.pl.cpdb_network(sub_adata,sub_i,celltype_key='cell_labels',
|
| 312 |
+
counts_min=10,
|
| 313 |
+
nodesize_scale=5,
|
| 314 |
+
ax=ax)
|
| 315 |
+
|
| 316 |
+
|
| 317 |
+
# In[21]:
|
| 318 |
+
|
| 319 |
+
|
| 320 |
+
fig, ax = plt.subplots(figsize=(3,3))
|
| 321 |
+
ov.pl.cpdb_heatmap(sub_adata,sub_i,celltype_key='cell_labels',
|
| 322 |
+
ax=ax,legend_kws={'fontsize':12,'bbox_to_anchor':(5, -0.9),'loc':'center left',})
|
| 323 |
+
|
| 324 |
+
|
| 325 |
+
# ## The ligand-receptor exacted
|
| 326 |
+
#
|
| 327 |
+
# We can set EVT as ligand or receptor to exacted the ligand-receptor proteins from the result of CellPhoneDB.
|
| 328 |
+
|
| 329 |
+
#
|
| 330 |
+
#
|
| 331 |
+
# The most important step is that we need to extract the results of the analysis with eEVT as the ligand, and here we use ov's function `ov.single.cpdb_exact_target`,`ov.single.cpdb_exact_source` to do this
|
| 332 |
+
|
| 333 |
+
# In[22]:
|
| 334 |
+
|
| 335 |
+
|
| 336 |
+
sub_means=ov.single.cpdb_exact_target(cpdb_results['means'],['eEVT','iEVT'])
|
| 337 |
+
sub_means=ov.single.cpdb_exact_source(sub_means,['dNK1','dNK2','dNK3'])
|
| 338 |
+
sub_means.head()
|
| 339 |
+
|
| 340 |
+
|
| 341 |
+
# In[23]:
|
| 342 |
+
|
| 343 |
+
|
| 344 |
+
ov.pl.cpdb_interacting_heatmap(adata=adata,
|
| 345 |
+
celltype_key='cell_labels',
|
| 346 |
+
means=cpdb_results['means'],
|
| 347 |
+
pvalues=cpdb_results['pvalues'],
|
| 348 |
+
source_cells=['dNK1','dNK2','dNK3'],
|
| 349 |
+
target_cells=['eEVT','iEVT'],
|
| 350 |
+
plot_secret=True,
|
| 351 |
+
min_means=3,
|
| 352 |
+
nodecolor_dict=None,
|
| 353 |
+
ax=None,
|
| 354 |
+
figsize=(2,6),
|
| 355 |
+
fontsize=10,)
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
# Sometimes we care about the expression of ligand in SOURCE and receptor in TARGET, we provide another function for getting the expression situation
|
| 359 |
+
|
| 360 |
+
# In[24]:
|
| 361 |
+
|
| 362 |
+
|
| 363 |
+
ov.pl.cpdb_group_heatmap(adata=adata,
|
| 364 |
+
celltype_key='cell_labels',
|
| 365 |
+
means=cpdb_results['means'],
|
| 366 |
+
cmap={'Target':'Blues','Source':'Reds'},
|
| 367 |
+
source_cells=['dNK1','dNK2','dNK3'],
|
| 368 |
+
target_cells=['eEVT','iEVT'],
|
| 369 |
+
plot_secret=True,
|
| 370 |
+
min_means=3,
|
| 371 |
+
nodecolor_dict=None,
|
| 372 |
+
ax=None,
|
| 373 |
+
figsize=(2,6),
|
| 374 |
+
fontsize=10,)
|
| 375 |
+
|
| 376 |
+
|
| 377 |
+
# We can also build Ligand, Receptor, SOURCE, and TARGET into a regulatory network, which is interesting.
|
| 378 |
+
|
| 379 |
+
# In[25]:
|
| 380 |
+
|
| 381 |
+
|
| 382 |
+
ov.pl.cpdb_interacting_network(adata=adata,
|
| 383 |
+
celltype_key='cell_labels',
|
| 384 |
+
means=cpdb_results['means'],
|
| 385 |
+
source_cells=['dNK1','dNK2','dNK3'],
|
| 386 |
+
target_cells=['eEVT','iEVT'],
|
| 387 |
+
means_min=1,
|
| 388 |
+
means_sum_min=1,
|
| 389 |
+
nodecolor_dict=None,
|
| 390 |
+
ax=None,
|
| 391 |
+
figsize=(6,6),
|
| 392 |
+
fontsize=10)
|
| 393 |
+
|
| 394 |
+
|
| 395 |
+
# Sometimes we want to analyse ligand-receptor pathway enrichment or function, so we need to extract ligand-receptor pairs from the significant ligand-receptors filtered out above, and omicverse provides an easy function `ov.single.cpdb_interaction_filtered` to do this here
|
| 396 |
+
|
| 397 |
+
# In[40]:
|
| 398 |
+
|
| 399 |
+
|
| 400 |
+
sub_means=sub_means.loc[~sub_means['gene_a'].isnull()]
|
| 401 |
+
sub_means=sub_means.loc[~sub_means['gene_b'].isnull()]
|
| 402 |
+
enrichr_genes=sub_means['gene_a'].tolist()+sub_means['gene_b'].tolist()
|
| 403 |
+
|
| 404 |
+
|
| 405 |
+
# A tutorial on enrichment you can find in the Bulk chapter of tutorials:
|
| 406 |
+
#
|
| 407 |
+
# https://omicverse.readthedocs.io/en/latest/Tutorials-bulk/t_deg/ or https://starlitnightly.github.io/omicverse/Tutorials-bulk/t_deg/
|
| 408 |
+
|
| 409 |
+
# In[ ]:
|
| 410 |
+
|
| 411 |
+
|
| 412 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Biological_Process_2023.txt',organism='Human')
|
| 413 |
+
|
| 414 |
+
|
| 415 |
+
# In[14]:
|
| 416 |
+
|
| 417 |
+
|
| 418 |
+
#deg_genes=dds.result.loc[dds.result['sig']!='normal'].index.tolist()
|
| 419 |
+
enr=ov.bulk.geneset_enrichment(gene_list=enrichr_genes,
|
| 420 |
+
pathways_dict=pathway_dict,
|
| 421 |
+
pvalue_type='auto',
|
| 422 |
+
organism='human')
|
| 423 |
+
|
| 424 |
+
|
| 425 |
+
# In[20]:
|
| 426 |
+
|
| 427 |
+
|
| 428 |
+
ov.plot_set()
|
| 429 |
+
ov.bulk.geneset_plot(enr,figsize=(2,4),fig_title='GO-Bio(EVT)',
|
| 430 |
+
cax_loc=[2, 0.45, 0.5, 0.02],num=8,
|
| 431 |
+
bbox_to_anchor_used=(-0.25, -13),custom_ticks=[10,100],
|
| 432 |
+
cmap='Greens')
|
| 433 |
+
|
| 434 |
+
|
| 435 |
+
# In[ ]:
|
| 436 |
+
|
| 437 |
+
|
| 438 |
+
|
| 439 |
+
|
ovrawm/t_cluster.txt
ADDED
|
@@ -0,0 +1,312 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Clustering space
|
| 5 |
+
#
|
| 6 |
+
# In this tutorial, we will explore how to run the Supervised clustering, unsupervised clustering, and amortized Latent Dirichlet Allocation (LDA) model implementation in `omicverse` with `GaussianMixture`,`Leiden/Louvain` and `MiRA`.
|
| 7 |
+
#
|
| 8 |
+
# In scRNA-seq data analysis, we describe cellular structure in our dataset with finding cell identities that relate to known cell states or cell cycle stages. This process is usually called cell identity annotation. For this purpose, we structure cells into clusters to infer the identity of similar cells. Clustering itself is a common unsupervised machine learning problem.
|
| 9 |
+
#
|
| 10 |
+
# LDA is a topic modelling method first introduced in the natural language processing field. By treating each cell as a document and each gene expression count as a word, we can carry over the method to the single-cell biology field.
|
| 11 |
+
#
|
| 12 |
+
# Below, we will train the model over a dataset, plot the topics over a UMAP of the reference set, and inspect the topics for characteristic gene sets.
|
| 13 |
+
#
|
| 14 |
+
# ## Preprocess data
|
| 15 |
+
#
|
| 16 |
+
# As an example, we apply differential kinetic analysis to dentate gyrus neurogenesis, which comprises multiple heterogeneous subpopulations.
|
| 17 |
+
#
|
| 18 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1d_szq-y-g7O0C5rJgK22XC7uWTUNrYpK?usp=sharing
|
| 19 |
+
|
| 20 |
+
# In[1]:
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
import omicverse as ov
|
| 24 |
+
import scanpy as sc
|
| 25 |
+
import scvelo as scv
|
| 26 |
+
ov.plot_set()
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
import scvelo as scv
|
| 33 |
+
adata=scv.datasets.dentategyrus()
|
| 34 |
+
adata
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
# In[3]:
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,)
|
| 41 |
+
adata.raw = adata
|
| 42 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 43 |
+
ov.pp.scale(adata)
|
| 44 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
# Let us inspect the contribution of single PCs to the total variance in the data. This gives us information about how many PCs we should consider in order to compute the neighborhood relations of cells. In our experience, often a rough estimate of the number of PCs does fine.
|
| 48 |
+
|
| 49 |
+
# In[4]:
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
ov.utils.plot_pca_variance_ratio(adata)
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
# ## Unsupervised clustering
|
| 56 |
+
#
|
| 57 |
+
# The Leiden algorithm is as an improved version of the Louvain algorithm which outperformed other clustering methods for single-cell RNA-seq data analysis ([Du et al., 2018, Freytag et al., 2018, Weber and Robinson, 2016]). Since the Louvain algorithm is no longer maintained, using Leiden instead is preferred.
|
| 58 |
+
#
|
| 59 |
+
# We, therefore, propose to use the Leiden algorithm[Traag et al., 2019] on single-cell k-nearest-neighbour (KNN) graphs to cluster single-cell datasets.
|
| 60 |
+
#
|
| 61 |
+
# Leiden creates clusters by taking into account the number of links between cells in a cluster versus the overall expected number of links in the dataset.
|
| 62 |
+
#
|
| 63 |
+
# Here, we set `method='leiden'` to cluster the cells using `Leiden`
|
| 64 |
+
#
|
| 65 |
+
|
| 66 |
+
# In[5]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 70 |
+
use_rep='scaled|original|X_pca')
|
| 71 |
+
ov.utils.cluster(adata,method='leiden',resolution=1)
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
# In[6]:
|
| 75 |
+
|
| 76 |
+
|
| 77 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 78 |
+
color=['clusters','leiden'],
|
| 79 |
+
frameon='small',wspace=0.5)
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
# We can also set `method='louvain'` to cluster the cells using `Louvain`
|
| 83 |
+
|
| 84 |
+
# In[7]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 88 |
+
use_rep='scaled|original|X_pca')
|
| 89 |
+
ov.utils.cluster(adata,method='louvain',resolution=1)
|
| 90 |
+
|
| 91 |
+
|
| 92 |
+
# In[8]:
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 96 |
+
color=['clusters','louvain'],
|
| 97 |
+
frameon='small',wspace=0.5)
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# ## Supervised clustering
|
| 101 |
+
#
|
| 102 |
+
# In addition to clustering using unsupervised clustering methods, we can also try supervised clustering methods, such as Gaussian mixture model clustering, which is a supervised clustering algorithm that works better in machine learning
|
| 103 |
+
#
|
| 104 |
+
# Gaussian mixture models can be used to cluster unlabeled data in much the same way as k-means. There are, however, a couple of advantages to using Gaussian mixture models over k-means.
|
| 105 |
+
#
|
| 106 |
+
# First and foremost, k-means does not account for variance. By variance, we are referring to the width of the bell shape curve.
|
| 107 |
+
#
|
| 108 |
+
# The second difference between k-means and Gaussian mixture models is that the former performs hard classification whereas the latter performs soft classification. In other words, k-means tells us what data point belong to which cluster but won’t provide us with the probabilities that a given data point belongs to each of the possible clusters.
|
| 109 |
+
#
|
| 110 |
+
# Here, we set `method='GMM'` to cluster the cells using `GaussianMixture`,`n_components` means the PCs to be used in clustering, `covariance_type` means the Gaussian Mixture Models (`diagonal`, `spherical`, `tied` and `full` covariance matrices supported). More arguments could found in [`sklearn.mixture.GaussianMixture`](http://scikit-learn.org/stable/modules/generated/sklearn.mixture.GaussianMixture.html)
|
| 111 |
+
#
|
| 112 |
+
|
| 113 |
+
# In[9]:
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
ov.utils.cluster(adata,use_rep='scaled|original|X_pca',
|
| 117 |
+
method='GMM',n_components=21,
|
| 118 |
+
covariance_type='full',tol=1e-9, max_iter=1000, )
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# In[10]:
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 125 |
+
color=['clusters','gmm_cluster'],
|
| 126 |
+
frameon='small',wspace=0.5)
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
# ## Latent Dirichlet Allocation (LDA) model implementation
|
| 130 |
+
#
|
| 131 |
+
# Topic models, like Latent Dirichlet Allocation (LDA), have traditionally been used to decompose a corpus of text into topics - or themes - composed of words that often appear together in documents. Documents, in turn, are modeled as a mixture of topics based on the words they contain.
|
| 132 |
+
#
|
| 133 |
+
# MIRA extends these ideas to single-cell genomics data, where topics are groups of genes that are co-expressed or cis-regulatory elements that are co-accessible, and cells are a mixture of these regulatory modules.
|
| 134 |
+
#
|
| 135 |
+
# Here, we used `ov.utils.LDA_topic` to construct the model of MiRA.
|
| 136 |
+
#
|
| 137 |
+
# Particularly, and at a minimum, we must tell the model
|
| 138 |
+
#
|
| 139 |
+
# - feature_type: what type of features we are working with (either “expression” or “accessibility”)
|
| 140 |
+
# - highly_variable_key: which .var key to find our highly variable genes
|
| 141 |
+
# - counts_layer: which layer to get the raw counts from.
|
| 142 |
+
# - categorical_covariates, continuous_covariates: Technical variables influencing the generative process of the data. For example, a categorical technical factor may be the cells’ batch of origin, as shown here. A continous technical factor might be % of mitchondrial reads. For unbatched data, ignore these parameters.
|
| 143 |
+
# - learning_rate: for larger datasets, the default of 1e-3, 0.1 usually works well.
|
| 144 |
+
|
| 145 |
+
# In[11]:
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
LDA_obj=ov.utils.LDA_topic(adata,feature_type='expression',
|
| 149 |
+
highly_variable_key='highly_variable_features',
|
| 150 |
+
layers='counts',batch_key=None,learning_rate=1e-3)
|
| 151 |
+
|
| 152 |
+
|
| 153 |
+
# This method works by instantiating a special version of the CODAL model with far too many topics, which are gradually pruned if that topic is not needed to describe the data. The function returns the maximum contribution of each topic to any cell in the dataset. The predicted number of topics is given by the elbo of the maximum contribution curve, minus 1. A rule of thumb is that the last valid topic to include in the model is followed by a drop-off, after which all subsequent topics hover between 0.-0.05 maximum contributions.
|
| 154 |
+
#
|
| 155 |
+
# We set `NUM_TOPICS` to six to try.
|
| 156 |
+
|
| 157 |
+
# In[12]:
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
LDA_obj.plot_topic_contributions(6)
|
| 161 |
+
|
| 162 |
+
|
| 163 |
+
# We can observe that there are 13 TOPICs to be above the threshold line, so we set the new NUM_TOPIC to 12
|
| 164 |
+
|
| 165 |
+
# In[13]:
|
| 166 |
+
|
| 167 |
+
|
| 168 |
+
LDA_obj.predicted(13)
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
# One can plot the distribution of topics across cells to see how the latent space reflects changes in cell state:
|
| 172 |
+
|
| 173 |
+
# In[14]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
ov.plot_set()
|
| 177 |
+
ov.utils.embedding(adata, basis='X_umap',color = LDA_obj.model.topic_cols, cmap='BuPu', ncols=4,
|
| 178 |
+
add_outline=True, frameon='small',)
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
# In[15]:
|
| 182 |
+
|
| 183 |
+
|
| 184 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 185 |
+
color=['clusters','LDA_cluster'],
|
| 186 |
+
frameon='small',wspace=0.5)
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# Here we are, proposing another idea of categorisation. We use cells with LDA greater than 0.4 as a primitive class, and then train a random forest classification model, and then use the random forest classification model to classify cells with LDA less than 0.5 to get a more accurate
|
| 190 |
+
|
| 191 |
+
# In[20]:
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
LDA_obj.get_results_rfc(adata,use_rep='scaled|original|X_pca',
|
| 195 |
+
LDA_threshold=0.4,num_topics=13)
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
# In[21]:
|
| 199 |
+
|
| 200 |
+
|
| 201 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 202 |
+
color=['LDA_cluster_rfc','LDA_cluster_clf'],
|
| 203 |
+
frameon='small',wspace=0.5)
|
| 204 |
+
|
| 205 |
+
|
| 206 |
+
# ## cNMF
|
| 207 |
+
#
|
| 208 |
+
# More detail could be found in https://starlitnightly.github.io/omicverse/Tutorials-single/t_cnmf/
|
| 209 |
+
|
| 210 |
+
# In[32]:
|
| 211 |
+
|
| 212 |
+
|
| 213 |
+
adata.X.toarray()
|
| 214 |
+
|
| 215 |
+
|
| 216 |
+
# In[ ]:
|
| 217 |
+
|
| 218 |
+
|
| 219 |
+
import numpy as np
|
| 220 |
+
## Initialize the cnmf object that will be used to run analyses
|
| 221 |
+
cnmf_obj = ov.single.cNMF(adata,components=np.arange(5,11), n_iter=20, seed=14, num_highvar_genes=2000,
|
| 222 |
+
output_dir='example_dg1/cNMF', name='dg_cNMF')
|
| 223 |
+
## Specify that the jobs are being distributed over a single worker (total_workers=1) and then launch that worker
|
| 224 |
+
cnmf_obj.factorize(worker_i=0, total_workers=4)
|
| 225 |
+
cnmf_obj.combine(skip_missing_files=True)
|
| 226 |
+
cnmf_obj.k_selection_plot(close_fig=False)
|
| 227 |
+
|
| 228 |
+
|
| 229 |
+
# In[35]:
|
| 230 |
+
|
| 231 |
+
|
| 232 |
+
selected_K = 7
|
| 233 |
+
density_threshold = 2.00
|
| 234 |
+
cnmf_obj.consensus(k=selected_K,
|
| 235 |
+
density_threshold=density_threshold,
|
| 236 |
+
show_clustering=True,
|
| 237 |
+
close_clustergram_fig=False)
|
| 238 |
+
result_dict = cnmf_obj.load_results(K=selected_K, density_threshold=density_threshold)
|
| 239 |
+
cnmf_obj.get_results(adata,result_dict)
|
| 240 |
+
|
| 241 |
+
|
| 242 |
+
# In[36]:
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
ov.pl.embedding(adata, basis='X_umap',color=result_dict['usage_norm'].columns,
|
| 246 |
+
use_raw=False, ncols=3, vmin=0, vmax=1,frameon='small')
|
| 247 |
+
|
| 248 |
+
|
| 249 |
+
# In[40]:
|
| 250 |
+
|
| 251 |
+
|
| 252 |
+
cnmf_obj.get_results_rfc(adata,result_dict,
|
| 253 |
+
use_rep='scaled|original|X_pca',
|
| 254 |
+
cNMF_threshold=0.5)
|
| 255 |
+
|
| 256 |
+
|
| 257 |
+
# In[41]:
|
| 258 |
+
|
| 259 |
+
|
| 260 |
+
ov.pl.embedding(
|
| 261 |
+
adata,
|
| 262 |
+
basis="X_umap",
|
| 263 |
+
color=['cNMF_cluster_rfc','cNMF_cluster_clf'],
|
| 264 |
+
frameon='small',
|
| 265 |
+
#title="Celltypes",
|
| 266 |
+
#legend_loc='on data',
|
| 267 |
+
legend_fontsize=14,
|
| 268 |
+
legend_fontoutline=2,
|
| 269 |
+
#size=10,
|
| 270 |
+
#legend_loc=True,
|
| 271 |
+
add_outline=False,
|
| 272 |
+
#add_outline=True,
|
| 273 |
+
outline_color='black',
|
| 274 |
+
outline_width=1,
|
| 275 |
+
show=False,
|
| 276 |
+
)
|
| 277 |
+
|
| 278 |
+
|
| 279 |
+
# ## Evaluation the clustering space
|
| 280 |
+
#
|
| 281 |
+
# Rand index adjusted for chance. The Rand Index computes a similarity measure between two clusterings by considering all pairs of samples and counting pairs that are assigned in the same or different clusters in the predicted and true clusterings.
|
| 282 |
+
|
| 283 |
+
# In[42]:
|
| 284 |
+
|
| 285 |
+
|
| 286 |
+
from sklearn.metrics.cluster import adjusted_rand_score
|
| 287 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['leiden'])
|
| 288 |
+
print('Leiden, Adjusted rand index = %.2f' %ARI)
|
| 289 |
+
|
| 290 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['louvain'])
|
| 291 |
+
print('Louvain, Adjusted rand index = %.2f' %ARI)
|
| 292 |
+
|
| 293 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['gmm_cluster'])
|
| 294 |
+
print('GMM, Adjusted rand index = %.2f' %ARI)
|
| 295 |
+
|
| 296 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['LDA_cluster'])
|
| 297 |
+
print('LDA, Adjusted rand index = %.2f' %ARI)
|
| 298 |
+
|
| 299 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['LDA_cluster_rfc'])
|
| 300 |
+
print('LDA_rfc, Adjusted rand index = %.2f' %ARI)
|
| 301 |
+
|
| 302 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['LDA_cluster_clf'])
|
| 303 |
+
print('LDA_clf, Adjusted rand index = %.2f' %ARI)
|
| 304 |
+
|
| 305 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['cNMF_cluster_rfc'])
|
| 306 |
+
print('cNMF_rfc, Adjusted rand index = %.2f' %ARI)
|
| 307 |
+
|
| 308 |
+
ARI = adjusted_rand_score(adata.obs['clusters'], adata.obs['cNMF_cluster_clf'])
|
| 309 |
+
print('cNMF_clf, Adjusted rand index = %.2f' %ARI)
|
| 310 |
+
|
| 311 |
+
|
| 312 |
+
# We can find that the LDA topic model is the most effective among the above clustering algorithms, but it also takes the longest computation time and requires GPU resources. We notice that the Gaussian mixture model is second only to the LDA topic model. The GMM will be a great choice in omicverse's future clustering algorithms for spatial transcriptomics.
|
ovrawm/t_cluster_space.txt
ADDED
|
@@ -0,0 +1,399 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Spatial clustering and denoising expressions
|
| 5 |
+
#
|
| 6 |
+
# Spatial clustering, which shares an analogy with single-cell clustering, has expanded the scope of tissue physiology studies from cell-centroid to structure-centroid with spatially resolved transcriptomics (SRT) data.
|
| 7 |
+
#
|
| 8 |
+
# Here, we presented four spatial clustering methods in OmicVerse.
|
| 9 |
+
#
|
| 10 |
+
# We made three improvements in integrating the `GraphST`,`BINARY`,`CAST` and `STAGATE` algorithm in OmicVerse:
|
| 11 |
+
# - We removed the preprocessing that comes with `GraphST` and used the preprocessing consistent with all SRTs in OmicVerse
|
| 12 |
+
# - We optimised the dimensional display of `GraphST`, and PCA is considered a self-contained computational step.
|
| 13 |
+
# - We implemented `mclust` using Python, removing the R language dependency.
|
| 14 |
+
# - We provided a unified interface `ov.space.cluster`, the user can use the function interface at once to complete all the simultaneous
|
| 15 |
+
#
|
| 16 |
+
# If you found this tutorial helpful, please cite `GraphST`,`BINARY`,`CAST` and `STAGATE` and `OmicVerse`:
|
| 17 |
+
#
|
| 18 |
+
# - Long, Y., Ang, K.S., Li, M. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat Commun 14, 1155 (2023). https://doi.org/10.1038/s41467-023-36796-3
|
| 19 |
+
# - Lin S, Cui Y, Zhao F, Yang Z, Song J, Yao J, et al. Complete spatially resolved gene expression is not necessary for identifying spatial domains. Cell Genomics. 2024;4:100565.
|
| 20 |
+
# - Tang, Z., Luo, S., Zeng, H. et al. Search and match across spatial omics samples at single-cell resolution. Nat Methods 21, 1818–1829 (2024). https://doi.org/10.1038/s41592-024-02410-7
|
| 21 |
+
# - Dong, K., Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 13, 1739 (2022). https://doi.org/10.1038/s41467-022-29439-6
|
| 22 |
+
#
|
| 23 |
+
#
|
| 24 |
+
|
| 25 |
+
# In[1]:
|
| 26 |
+
|
| 27 |
+
|
| 28 |
+
import omicverse as ov
|
| 29 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 30 |
+
import scanpy as sc
|
| 31 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 32 |
+
ov.plot_set()
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# ## Preprocess data
|
| 36 |
+
#
|
| 37 |
+
# Here we present our re-analysis of 151676 sample of the dorsolateral prefrontal cortex (DLPFC) dataset. Maynard et al. has manually annotated DLPFC layers and white matter (WM) based on the morphological features and gene markers.
|
| 38 |
+
#
|
| 39 |
+
# This tutorial demonstrates how to identify spatial domains on 10x Visium data using STAGATE. The processed data are available at https://github.com/LieberInstitute/spatialLIBD. We downloaded the manual annotation from the spatialLIBD package and provided at https://drive.google.com/drive/folders/10lhz5VY7YfvHrtV40MwaqLmWz56U9eBP?usp=sharing.
|
| 40 |
+
|
| 41 |
+
# In[2]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
adata = sc.read_visium(path='data', count_file='151676_filtered_feature_bc_matrix.h5')
|
| 45 |
+
adata.var_names_make_unique()
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# <div class="admonition warning">
|
| 49 |
+
# <p class="admonition-title">Note</p>
|
| 50 |
+
# <p>
|
| 51 |
+
# We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code.
|
| 52 |
+
# </p>
|
| 53 |
+
# </div>
|
| 54 |
+
#
|
| 55 |
+
# ```python
|
| 56 |
+
# #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 57 |
+
# #adata.raw = adata
|
| 58 |
+
# #adata = adata[:, adata.var.highly_variable_features]
|
| 59 |
+
# ```
|
| 60 |
+
|
| 61 |
+
# In[3]:
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
sc.pp.calculate_qc_metrics(adata, inplace=True)
|
| 65 |
+
adata = adata[:,adata.var['total_counts']>100]
|
| 66 |
+
adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",)
|
| 67 |
+
adata
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
# In[5]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
adata.write('data/cluster_svg.h5ad',compression='gzip')
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
# In[2]:
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
adata=ov.read('data/cluster_svg.h5ad',compression='gzip')
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
# (Optional) We read the ground truth area of our spatial data
|
| 83 |
+
#
|
| 84 |
+
# This step is not mandatory to run, in the tutorial, it's just to demonstrate the accuracy of our clustering effect, and in your own tasks, there is often no Ground_truth
|
| 85 |
+
|
| 86 |
+
# In[3]:
|
| 87 |
+
|
| 88 |
+
|
| 89 |
+
# read the annotation
|
| 90 |
+
import pandas as pd
|
| 91 |
+
import os
|
| 92 |
+
Ann_df = pd.read_csv(os.path.join('data', '151676_truth.txt'), sep='\t', header=None, index_col=0)
|
| 93 |
+
Ann_df.columns = ['Ground Truth']
|
| 94 |
+
adata.obs['Ground Truth'] = Ann_df.loc[adata.obs_names, 'Ground Truth']
|
| 95 |
+
sc.pl.spatial(adata, img_key="hires", color=["Ground Truth"])
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# ## Method1: GraphST
|
| 99 |
+
#
|
| 100 |
+
# GraphST was rated as one of the best spatial clustering algorithms on Nature Method 2024.04, so we first tried to call GraphST for spatial domain identification in OmicVerse.
|
| 101 |
+
|
| 102 |
+
# In[4]:
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
methods_kwargs={}
|
| 106 |
+
methods_kwargs['GraphST']={
|
| 107 |
+
'device':'cuda:0',
|
| 108 |
+
'n_pcs':30
|
| 109 |
+
}
|
| 110 |
+
|
| 111 |
+
adata=ov.space.clusters(adata,
|
| 112 |
+
methods=['GraphST'],
|
| 113 |
+
methods_kwargs=methods_kwargs,
|
| 114 |
+
lognorm=1e4)
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
# In[11]:
|
| 118 |
+
|
| 119 |
+
|
| 120 |
+
ov.utils.cluster(adata,use_rep='graphst|original|X_pca',method='mclust',n_components=10,
|
| 121 |
+
modelNames='EEV', random_state=112,
|
| 122 |
+
)
|
| 123 |
+
adata.obs['mclust_GraphST'] = ov.utils.refine_label(adata, radius=50, key='mclust')
|
| 124 |
+
adata.obs['mclust_GraphST']=adata.obs['mclust_GraphST'].astype('category')
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
# In[12]:
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
res=ov.space.merge_cluster(adata,groupby='mclust_GraphST',use_rep='graphst|original|X_pca',
|
| 131 |
+
threshold=0.2,plot=True)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# In[13]:
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
sc.pl.spatial(adata, color=['mclust_GraphST','mclust_GraphST_tree','mclust','Ground Truth'])
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
# We can also use `mclust_R` to cluster the spatial domain, but this method need to install `rpy2` at first.
|
| 141 |
+
#
|
| 142 |
+
# The use of the mclust algorithm requires the rpy2 package and the mclust package. See https://pypi.org/project/rpy2/ and https://cran.r-project.org/web/packages/mclust/index.html for detail.
|
| 143 |
+
|
| 144 |
+
# In[14]:
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
ov.utils.cluster(adata,use_rep='graphst|original|X_pca',method='mclust_R',n_components=10,
|
| 148 |
+
random_state=42,
|
| 149 |
+
)
|
| 150 |
+
adata.obs['mclust_R_GraphST'] = ov.utils.refine_label(adata, radius=30, key='mclust_R')
|
| 151 |
+
adata.obs['mclust_R_GraphST']=adata.obs['mclust_R_GraphST'].astype('category')
|
| 152 |
+
res=ov.space.merge_cluster(adata,groupby='mclust_R_GraphST',use_rep='graphst|original|X_pca',
|
| 153 |
+
threshold=0.2,plot=True)
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# In[15]:
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
sc.pl.spatial(adata, color=['mclust_R_GraphST','mclust_R_GraphST_tree','mclust','Ground Truth'])
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
# ## Method2: BINARY
|
| 163 |
+
#
|
| 164 |
+
# BINARY outperforms existing methods across various SRT data types while using significantly less input information.
|
| 165 |
+
#
|
| 166 |
+
# If your data is very large, or very sparse, I believe BINARY would be a great choice.
|
| 167 |
+
|
| 168 |
+
# In[3]:
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
methods_kwargs={}
|
| 172 |
+
methods_kwargs['BINARY']={
|
| 173 |
+
'use_method':'KNN',
|
| 174 |
+
'cutoff':6,
|
| 175 |
+
'obs_key':'BINARY_sample',
|
| 176 |
+
'use_list':None,
|
| 177 |
+
'pos_weight':10,
|
| 178 |
+
'device':'cuda:0',
|
| 179 |
+
'hidden_dims':[512, 30],
|
| 180 |
+
'n_epochs': 1000,
|
| 181 |
+
'lr': 0.001,
|
| 182 |
+
'key_added': 'BINARY',
|
| 183 |
+
'gradient_clipping': 5,
|
| 184 |
+
'weight_decay': 0.0001,
|
| 185 |
+
'verbose': True,
|
| 186 |
+
'random_seed':0,
|
| 187 |
+
'lognorm':1e4,
|
| 188 |
+
'n_top_genes':2000,
|
| 189 |
+
}
|
| 190 |
+
adata=ov.space.clusters(adata,
|
| 191 |
+
methods=['BINARY'],
|
| 192 |
+
methods_kwargs=methods_kwargs)
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
# if you want to use R's `mclust`, you can use `ov.utils.cluster`.
|
| 196 |
+
#
|
| 197 |
+
# But you need to install `rpy2` and `mclust` at first.
|
| 198 |
+
|
| 199 |
+
# In[4]:
|
| 200 |
+
|
| 201 |
+
|
| 202 |
+
ov.utils.cluster(adata,use_rep='BINARY',method='mclust_R',n_components=10,
|
| 203 |
+
random_state=42,
|
| 204 |
+
)
|
| 205 |
+
adata.obs['mclust_BINARY'] = ov.utils.refine_label(adata, radius=30, key='mclust_R')
|
| 206 |
+
adata.obs['mclust_BINARY']=adata.obs['mclust_BINARY'].astype('category')
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
# In[5]:
|
| 210 |
+
|
| 211 |
+
|
| 212 |
+
res=ov.space.merge_cluster(adata,groupby='mclust_BINARY',use_rep='BINARY',
|
| 213 |
+
threshold=0.01,plot=True)
|
| 214 |
+
|
| 215 |
+
|
| 216 |
+
# In[6]:
|
| 217 |
+
|
| 218 |
+
|
| 219 |
+
sc.pl.spatial(adata, color=['mclust_BINARY','mclust_BINARY_tree','mclust','Ground Truth'])
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
# In[10]:
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
ov.utils.cluster(adata,use_rep='BINARY',method='mclust',n_components=10,
|
| 226 |
+
modelNames='EEV', random_state=42,
|
| 227 |
+
)
|
| 228 |
+
adata.obs['mclustpy_BINARY'] = ov.utils.refine_label(adata, radius=30, key='mclust')
|
| 229 |
+
adata.obs['mclustpy_BINARY']=adata.obs['mclustpy_BINARY'].astype('category')
|
| 230 |
+
|
| 231 |
+
|
| 232 |
+
# In[13]:
|
| 233 |
+
|
| 234 |
+
|
| 235 |
+
adata.obs['mclustpy_BINARY']=adata.obs['mclustpy_BINARY'].astype('category')
|
| 236 |
+
res=ov.space.merge_cluster(adata,groupby='mclustpy_BINARY',use_rep='BINARY',
|
| 237 |
+
threshold=0.013,plot=True)
|
| 238 |
+
|
| 239 |
+
|
| 240 |
+
# In[14]:
|
| 241 |
+
|
| 242 |
+
|
| 243 |
+
sc.pl.spatial(adata, color=['mclustpy_BINARY','mclustpy_BINARY_tree','mclust','Ground Truth'])
|
| 244 |
+
#adata.obs['mclust_BINARY'] = ov.utils.refine_label(adata, radius=30, key='mclust')
|
| 245 |
+
#adata.obs['mclust_BINARY']=adata.obs['mclust_BINARY'].astype('category')
|
| 246 |
+
|
| 247 |
+
|
| 248 |
+
# ## Method3: STAGATE
|
| 249 |
+
#
|
| 250 |
+
# STAGATE is designed for spatial clustering and denoising expressions of spatial resolved transcriptomics (ST) data.
|
| 251 |
+
#
|
| 252 |
+
# STAGATE learns low-dimensional latent embeddings with both spatial information and gene expressions via a graph attention auto-encoder. The method adopts an attention mechanism in the middle layer of the encoder and decoder, which adaptively learns the edge weights of spatial neighbor networks, and further uses them to update the spot representation by collectively aggregating information from its neighbors. The latent embeddings and the reconstructed expression profiles can be used to downstream tasks such as spatial domain identification, visualization, spatial trajectory inference, data denoising and 3D expression domain extraction.
|
| 253 |
+
#
|
| 254 |
+
# Dong, Kangning, and Shihua Zhang. “Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder.” Nature Communications 13.1 (2022): 1-12.
|
| 255 |
+
#
|
| 256 |
+
#
|
| 257 |
+
# Here, we used `ov.space.pySTAGATE` to construct a STAGATE object to train the model.
|
| 258 |
+
#
|
| 259 |
+
|
| 260 |
+
# In[12]:
|
| 261 |
+
|
| 262 |
+
|
| 263 |
+
methods_kwargs={}
|
| 264 |
+
methods_kwargs['STAGATE']={
|
| 265 |
+
'num_batch_x':3,'num_batch_y':2,
|
| 266 |
+
'spatial_key':['X','Y'],'rad_cutoff':200,
|
| 267 |
+
'num_epoch':1000,'lr':0.001,
|
| 268 |
+
'weight_decay':1e-4,'hidden_dims':[512, 30],
|
| 269 |
+
'device':'cuda:0',
|
| 270 |
+
#'n_top_genes':2000,
|
| 271 |
+
}
|
| 272 |
+
|
| 273 |
+
adata=ov.space.clusters(adata,
|
| 274 |
+
methods=['STAGATE'],
|
| 275 |
+
methods_kwargs=methods_kwargs)
|
| 276 |
+
|
| 277 |
+
|
| 278 |
+
# In[36]:
|
| 279 |
+
|
| 280 |
+
|
| 281 |
+
ov.utils.cluster(adata,use_rep='STAGATE',method='mclust_R',n_components=10,
|
| 282 |
+
random_state=112,
|
| 283 |
+
)
|
| 284 |
+
adata.obs['mclust_R_STAGATE'] = ov.utils.refine_label(adata, radius=30, key='mclust_R')
|
| 285 |
+
adata.obs['mclust_R_STAGATE']=adata.obs['mclust_R_STAGATE'].astype('category')
|
| 286 |
+
res=ov.space.merge_cluster(adata,groupby='mclust_R_STAGATE',use_rep='STAGATE',
|
| 287 |
+
threshold=0.005,plot=True)
|
| 288 |
+
|
| 289 |
+
|
| 290 |
+
# In[37]:
|
| 291 |
+
|
| 292 |
+
|
| 293 |
+
sc.pl.spatial(adata, color=['mclust_R_STAGATE','mclust_R_STAGATE_tree','mclust_R','Ground Truth'])
|
| 294 |
+
|
| 295 |
+
|
| 296 |
+
# ### Denoising
|
| 297 |
+
|
| 298 |
+
# In[52]:
|
| 299 |
+
|
| 300 |
+
|
| 301 |
+
adata.var.sort_values('PI',ascending=False).head(5)
|
| 302 |
+
|
| 303 |
+
|
| 304 |
+
# In[53]:
|
| 305 |
+
|
| 306 |
+
|
| 307 |
+
plot_gene = 'MBP'
|
| 308 |
+
import matplotlib.pyplot as plt
|
| 309 |
+
fig, axs = plt.subplots(1, 2, figsize=(8, 4))
|
| 310 |
+
sc.pl.spatial(adata, img_key="hires", color=plot_gene, show=False, ax=axs[0], title='RAW_'+plot_gene, vmax='p99')
|
| 311 |
+
sc.pl.spatial(adata, img_key="hires", color=plot_gene, show=False, ax=axs[1], title='STAGATE_'+plot_gene, layer='STAGATE_ReX', vmax='p99')
|
| 312 |
+
|
| 313 |
+
|
| 314 |
+
# ## Method4: CAST
|
| 315 |
+
#
|
| 316 |
+
# CAST would be a great algorithm if your spatial transcriptome is at single-cell resolution and in multiple slices.
|
| 317 |
+
|
| 318 |
+
# In[38]:
|
| 319 |
+
|
| 320 |
+
|
| 321 |
+
methods_kwargs={}
|
| 322 |
+
methods_kwargs['CAST']={
|
| 323 |
+
'output_path_t':'result/CAST_gas/output',
|
| 324 |
+
'device':'cuda:0',
|
| 325 |
+
'gpu_t':0
|
| 326 |
+
}
|
| 327 |
+
adata=ov.space.clusters(adata,
|
| 328 |
+
methods=['CAST'],
|
| 329 |
+
methods_kwargs=methods_kwargs)
|
| 330 |
+
|
| 331 |
+
|
| 332 |
+
# In[39]:
|
| 333 |
+
|
| 334 |
+
|
| 335 |
+
ov.utils.cluster(adata,use_rep='X_cast',method='mclust',n_components=10,
|
| 336 |
+
modelNames='EEV', random_state=42,
|
| 337 |
+
)
|
| 338 |
+
adata.obs['mclust_CAST'] = ov.utils.refine_label(adata, radius=50, key='mclust')
|
| 339 |
+
adata.obs['mclust_CAST']=adata.obs['mclust_CAST'].astype('category')
|
| 340 |
+
|
| 341 |
+
|
| 342 |
+
# In[40]:
|
| 343 |
+
|
| 344 |
+
|
| 345 |
+
res=ov.space.merge_cluster(adata,groupby='mclust_CAST',use_rep='X_cast',
|
| 346 |
+
threshold=0.1,plot=True)
|
| 347 |
+
|
| 348 |
+
|
| 349 |
+
# In[41]:
|
| 350 |
+
|
| 351 |
+
|
| 352 |
+
sc.pl.spatial(adata, color=['mclust_CAST','mclust_CAST_tree','mclust','Ground Truth'])
|
| 353 |
+
|
| 354 |
+
|
| 355 |
+
# In[42]:
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
adata
|
| 359 |
+
|
| 360 |
+
|
| 361 |
+
# ## Evaluate cluster
|
| 362 |
+
#
|
| 363 |
+
# We use ARI to evaluate the scoring of our clusters against the true values
|
| 364 |
+
#
|
| 365 |
+
# While it appears that STAGATE works best, note that this is only on this dataset.
|
| 366 |
+
# - If your data is spot-level resolution, GraphST, BINARY and STAGATE would be good algorithms to use
|
| 367 |
+
# - BINARY and CAST would be good algorithms if your data is NanoString or other single-cell resolution
|
| 368 |
+
|
| 369 |
+
# In[50]:
|
| 370 |
+
|
| 371 |
+
|
| 372 |
+
from sklearn.metrics.cluster import adjusted_rand_score
|
| 373 |
+
|
| 374 |
+
obs_df = adata.obs.dropna()
|
| 375 |
+
#GraphST
|
| 376 |
+
ARI = adjusted_rand_score(obs_df['mclust_GraphST'], obs_df['Ground Truth'])
|
| 377 |
+
print('mclust_GraphST: Adjusted rand index = %.2f' %ARI)
|
| 378 |
+
|
| 379 |
+
ARI = adjusted_rand_score(obs_df['mclust_R_GraphST'], obs_df['Ground Truth'])
|
| 380 |
+
print('mclust_R_GraphST: Adjusted rand index = %.2f' %ARI)
|
| 381 |
+
|
| 382 |
+
ARI = adjusted_rand_score(obs_df['mclust_R_STAGATE'], obs_df['Ground Truth'])
|
| 383 |
+
print('mclust_STAGATE: Adjusted rand index = %.2f' %ARI)
|
| 384 |
+
|
| 385 |
+
ARI = adjusted_rand_score(obs_df['mclust_BINARY'], obs_df['Ground Truth'])
|
| 386 |
+
print('mclust_BINARY: Adjusted rand index = %.2f' %ARI)
|
| 387 |
+
|
| 388 |
+
ARI = adjusted_rand_score(obs_df['mclustpy_BINARY'], obs_df['Ground Truth'])
|
| 389 |
+
print('mclustpy_BINARY: Adjusted rand index = %.2f' %ARI)
|
| 390 |
+
|
| 391 |
+
ARI = adjusted_rand_score(obs_df['mclust_CAST'], obs_df['Ground Truth'])
|
| 392 |
+
print('mclust_CAST: Adjusted rand index = %.2f' %ARI)
|
| 393 |
+
|
| 394 |
+
|
| 395 |
+
# In[ ]:
|
| 396 |
+
|
| 397 |
+
|
| 398 |
+
|
| 399 |
+
|
ovrawm/t_cnmf.txt
ADDED
|
@@ -0,0 +1,331 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Consensus Non-negative Matrix factorization (cNMF)
|
| 5 |
+
#
|
| 6 |
+
# cNMF is an analysis pipeline for inferring gene expression programs from single-cell RNA-Seq (scRNA-Seq) data.
|
| 7 |
+
#
|
| 8 |
+
# It takes a count matrix (N cells X G genes) as input and produces a (K x G) matrix of gene expression programs (GEPs) and a (N x K) matrix specifying the usage of each program for each cell in the data. You can read more about the method in the [github](https://github.com/dylkot/cNMF) and check out examples on dentategyrus.
|
| 9 |
+
|
| 10 |
+
# In[1]:
|
| 11 |
+
|
| 12 |
+
|
| 13 |
+
import scanpy as sc
|
| 14 |
+
import omicverse as ov
|
| 15 |
+
ov.plot_set()
|
| 16 |
+
|
| 17 |
+
|
| 18 |
+
# ## Loading dataset
|
| 19 |
+
#
|
| 20 |
+
# Here, we use the dentategyrus dataset as an example for cNMF.
|
| 21 |
+
|
| 22 |
+
# In[2]:
|
| 23 |
+
|
| 24 |
+
|
| 25 |
+
import scvelo as scv
|
| 26 |
+
adata=scv.datasets.dentategyrus()
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
# In[3]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)\nadata\n")
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# In[23]:
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
ov.pp.scale(adata)
|
| 39 |
+
ov.pp.pca(adata)
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
# In[4]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
import matplotlib.pyplot as plt
|
| 46 |
+
from matplotlib import patheffects
|
| 47 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 48 |
+
ov.pl.embedding(
|
| 49 |
+
adata,
|
| 50 |
+
basis="X_umap",
|
| 51 |
+
color=['clusters'],
|
| 52 |
+
frameon='small',
|
| 53 |
+
title="Celltypes",
|
| 54 |
+
#legend_loc='on data',
|
| 55 |
+
legend_fontsize=14,
|
| 56 |
+
legend_fontoutline=2,
|
| 57 |
+
#size=10,
|
| 58 |
+
ax=ax,
|
| 59 |
+
#legend_loc=True,
|
| 60 |
+
add_outline=False,
|
| 61 |
+
#add_outline=True,
|
| 62 |
+
outline_color='black',
|
| 63 |
+
outline_width=1,
|
| 64 |
+
show=False,
|
| 65 |
+
)
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
# ## Initialize and Training model
|
| 69 |
+
|
| 70 |
+
# In[5]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
import numpy as np
|
| 74 |
+
## Initialize the cnmf object that will be used to run analyses
|
| 75 |
+
cnmf_obj = ov.single.cNMF(adata,components=np.arange(5,11), n_iter=20, seed=14, num_highvar_genes=2000,
|
| 76 |
+
output_dir='example_dg/cNMF', name='dg_cNMF')
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# In[6]:
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
## Specify that the jobs are being distributed over a single worker (total_workers=1) and then launch that worker
|
| 83 |
+
cnmf_obj.factorize(worker_i=0, total_workers=2)
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# In[7]:
|
| 87 |
+
|
| 88 |
+
|
| 89 |
+
cnmf_obj.combine(skip_missing_files=True)
|
| 90 |
+
|
| 91 |
+
|
| 92 |
+
# ## Compute the stability and error at each choice of K to see if a clear choice jumps out.
|
| 93 |
+
#
|
| 94 |
+
# Please note that the maximum stability solution is not always the best choice depending on the application. However it is often a good starting point even if you have to investigate several choices of K
|
| 95 |
+
|
| 96 |
+
# In[8]:
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
cnmf_obj.k_selection_plot(close_fig=False)
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
# In this range, K=7 gave the most stable solution so we will begin by looking at that.
|
| 103 |
+
#
|
| 104 |
+
# The next step computes the consensus solution for a given choice of K. We first run it without any outlier filtering to see what that looks like. Setting the density threshold to anything >= 2.00 (the maximum possible distance between two unit vectors) ensures that nothing will be filtered.
|
| 105 |
+
#
|
| 106 |
+
# Then we run the consensus with a filter for outliers determined based on inspecting the histogram of distances between components and their nearest neighbors
|
| 107 |
+
|
| 108 |
+
# In[9]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
selected_K = 7
|
| 112 |
+
density_threshold = 2.00
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# In[10]:
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
cnmf_obj.consensus(k=selected_K,
|
| 119 |
+
density_threshold=density_threshold,
|
| 120 |
+
show_clustering=True,
|
| 121 |
+
close_clustergram_fig=False)
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# The above consensus plot shows that there is a substantial degree of concordance between the replicates with a few outliers. An outlier threshold of 0.1 seems appropriate
|
| 125 |
+
|
| 126 |
+
# In[11]:
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
density_threshold = 0.10
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# In[12]:
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
cnmf_obj.consensus(k=selected_K,
|
| 136 |
+
density_threshold=density_threshold,
|
| 137 |
+
show_clustering=True,
|
| 138 |
+
close_clustergram_fig=False)
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
# ## Visualization the result
|
| 142 |
+
|
| 143 |
+
# In[13]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
import seaborn as sns
|
| 147 |
+
import matplotlib.pyplot as plt
|
| 148 |
+
from matplotlib import patheffects
|
| 149 |
+
|
| 150 |
+
from matplotlib import gridspec
|
| 151 |
+
import matplotlib.pyplot as plt
|
| 152 |
+
|
| 153 |
+
width_ratios = [0.2, 4, 0.5, 10, 1]
|
| 154 |
+
height_ratios = [0.2, 4]
|
| 155 |
+
fig = plt.figure(figsize=(sum(width_ratios), sum(height_ratios)))
|
| 156 |
+
gs = gridspec.GridSpec(len(height_ratios), len(width_ratios), fig,
|
| 157 |
+
0.01, 0.01, 0.98, 0.98,
|
| 158 |
+
height_ratios=height_ratios,
|
| 159 |
+
width_ratios=width_ratios,
|
| 160 |
+
wspace=0, hspace=0)
|
| 161 |
+
|
| 162 |
+
D = cnmf_obj.topic_dist[cnmf_obj.spectra_order, :][:, cnmf_obj.spectra_order]
|
| 163 |
+
dist_ax = fig.add_subplot(gs[1,1], xscale='linear', yscale='linear',
|
| 164 |
+
xticks=[], yticks=[],xlabel='', ylabel='',
|
| 165 |
+
frameon=True)
|
| 166 |
+
dist_im = dist_ax.imshow(D, interpolation='none', cmap='viridis',
|
| 167 |
+
aspect='auto', rasterized=True)
|
| 168 |
+
|
| 169 |
+
left_ax = fig.add_subplot(gs[1,0], xscale='linear', yscale='linear', xticks=[], yticks=[],
|
| 170 |
+
xlabel='', ylabel='', frameon=True)
|
| 171 |
+
left_ax.imshow(cnmf_obj.kmeans_cluster_labels.values[cnmf_obj.spectra_order].reshape(-1, 1),
|
| 172 |
+
interpolation='none', cmap='Spectral', aspect='auto',
|
| 173 |
+
rasterized=True)
|
| 174 |
+
|
| 175 |
+
top_ax = fig.add_subplot(gs[0,1], xscale='linear', yscale='linear', xticks=[], yticks=[],
|
| 176 |
+
xlabel='', ylabel='', frameon=True)
|
| 177 |
+
top_ax.imshow(cnmf_obj.kmeans_cluster_labels.values[cnmf_obj.spectra_order].reshape(1, -1),
|
| 178 |
+
interpolation='none', cmap='Spectral', aspect='auto',
|
| 179 |
+
rasterized=True)
|
| 180 |
+
|
| 181 |
+
cbar_gs = gridspec.GridSpecFromSubplotSpec(3, 3, subplot_spec=gs[1, 2],
|
| 182 |
+
wspace=0, hspace=0)
|
| 183 |
+
cbar_ax = fig.add_subplot(cbar_gs[1,2], xscale='linear', yscale='linear',
|
| 184 |
+
xlabel='', ylabel='', frameon=True, title='Euclidean\nDistance')
|
| 185 |
+
cbar_ax.set_title('Euclidean\nDistance',fontsize=12)
|
| 186 |
+
vmin = D.min().min()
|
| 187 |
+
vmax = D.max().max()
|
| 188 |
+
fig.colorbar(dist_im, cax=cbar_ax,
|
| 189 |
+
ticks=np.linspace(vmin, vmax, 3),
|
| 190 |
+
)
|
| 191 |
+
cbar_ax.set_yticklabels(cbar_ax.get_yticklabels(),fontsize=12)
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
# In[14]:
|
| 195 |
+
|
| 196 |
+
|
| 197 |
+
density_filter = cnmf_obj.local_density.iloc[:, 0] < density_threshold
|
| 198 |
+
fig, hist_ax = plt.subplots(figsize=(4,4))
|
| 199 |
+
|
| 200 |
+
#hist_ax = fig.add_subplot(hist_gs[0,0], xscale='linear', yscale='linear',
|
| 201 |
+
# xlabel='', ylabel='', frameon=True, title='Local density histogram')
|
| 202 |
+
hist_ax.hist(cnmf_obj.local_density.values, bins=np.linspace(0, 1, 50))
|
| 203 |
+
hist_ax.yaxis.tick_right()
|
| 204 |
+
|
| 205 |
+
xlim = hist_ax.get_xlim()
|
| 206 |
+
ylim = hist_ax.get_ylim()
|
| 207 |
+
if density_threshold < xlim[1]:
|
| 208 |
+
hist_ax.axvline(density_threshold, linestyle='--', color='k')
|
| 209 |
+
hist_ax.text(density_threshold + 0.02, ylim[1] * 0.95, 'filtering\nthreshold\n\n', va='top')
|
| 210 |
+
hist_ax.set_xlim(xlim)
|
| 211 |
+
hist_ax.set_xlabel('Mean distance to k nearest neighbors\n\n%d/%d (%.0f%%) spectra above threshold\nwere removed prior to clustering'%(sum(~density_filter), len(density_filter), 100*(~density_filter).mean()))
|
| 212 |
+
hist_ax.set_title('Local density histogram')
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
# ## Explode the cNMF result
|
| 216 |
+
#
|
| 217 |
+
# We can load the results for a cNMF run with a given K and density filtering threshold like below
|
| 218 |
+
|
| 219 |
+
# In[15]:
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
result_dict = cnmf_obj.load_results(K=selected_K, density_threshold=density_threshold)
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
# In[16]:
|
| 226 |
+
|
| 227 |
+
|
| 228 |
+
result_dict['usage_norm'].head()
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
# In[17]:
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
result_dict['gep_scores'].head()
|
| 235 |
+
|
| 236 |
+
|
| 237 |
+
# In[18]:
|
| 238 |
+
|
| 239 |
+
|
| 240 |
+
result_dict['gep_tpm'].head()
|
| 241 |
+
|
| 242 |
+
|
| 243 |
+
# In[19]:
|
| 244 |
+
|
| 245 |
+
|
| 246 |
+
result_dict['top_genes'].head()
|
| 247 |
+
|
| 248 |
+
|
| 249 |
+
# We can extract cell classes directly based on the highest cNMF in each cell, but this has the disadvantage that it will lead to mixed cell classes if the heterogeneity of our data is not as strong as it should be.
|
| 250 |
+
|
| 251 |
+
# In[20]:
|
| 252 |
+
|
| 253 |
+
|
| 254 |
+
cnmf_obj.get_results(adata,result_dict)
|
| 255 |
+
|
| 256 |
+
|
| 257 |
+
# In[21]:
|
| 258 |
+
|
| 259 |
+
|
| 260 |
+
ov.pl.embedding(adata, basis='X_umap',color=result_dict['usage_norm'].columns,
|
| 261 |
+
use_raw=False, ncols=3, vmin=0, vmax=1,frameon='small')
|
| 262 |
+
|
| 263 |
+
|
| 264 |
+
# In[24]:
|
| 265 |
+
|
| 266 |
+
|
| 267 |
+
ov.pl.embedding(
|
| 268 |
+
adata,
|
| 269 |
+
basis="X_umap",
|
| 270 |
+
color=['cNMF_cluster'],
|
| 271 |
+
frameon='small',
|
| 272 |
+
#title="Celltypes",
|
| 273 |
+
#legend_loc='on data',
|
| 274 |
+
legend_fontsize=14,
|
| 275 |
+
legend_fontoutline=2,
|
| 276 |
+
#size=10,
|
| 277 |
+
#legend_loc=True,
|
| 278 |
+
add_outline=False,
|
| 279 |
+
#add_outline=True,
|
| 280 |
+
outline_color='black',
|
| 281 |
+
outline_width=1,
|
| 282 |
+
show=False,
|
| 283 |
+
)
|
| 284 |
+
|
| 285 |
+
|
| 286 |
+
# Here we are, proposing another idea of categorisation. We use cells with cNMF greater than 0.5 as a primitive class, and then train a random forest classification model, and then use the random forest classification model to classify cells with cNMF less than 0.5 to get a more accurate
|
| 287 |
+
|
| 288 |
+
# In[25]:
|
| 289 |
+
|
| 290 |
+
|
| 291 |
+
cnmf_obj.get_results_rfc(adata,result_dict,
|
| 292 |
+
use_rep='scaled|original|X_pca',
|
| 293 |
+
cNMF_threshold=0.5)
|
| 294 |
+
|
| 295 |
+
|
| 296 |
+
# In[27]:
|
| 297 |
+
|
| 298 |
+
|
| 299 |
+
ov.pl.embedding(
|
| 300 |
+
adata,
|
| 301 |
+
basis="X_umap",
|
| 302 |
+
color=['cNMF_cluster_rfc','cNMF_cluster_clf'],
|
| 303 |
+
frameon='small',
|
| 304 |
+
#title="Celltypes",
|
| 305 |
+
#legend_loc='on data',
|
| 306 |
+
legend_fontsize=14,
|
| 307 |
+
legend_fontoutline=2,
|
| 308 |
+
#size=10,
|
| 309 |
+
#legend_loc=True,
|
| 310 |
+
add_outline=False,
|
| 311 |
+
#add_outline=True,
|
| 312 |
+
outline_color='black',
|
| 313 |
+
outline_width=1,
|
| 314 |
+
show=False,
|
| 315 |
+
)
|
| 316 |
+
|
| 317 |
+
|
| 318 |
+
# In[25]:
|
| 319 |
+
|
| 320 |
+
|
| 321 |
+
plot_genes=[]
|
| 322 |
+
for i in result_dict['top_genes'].columns:
|
| 323 |
+
plot_genes+=result_dict['top_genes'][i][:3].values.reshape(-1).tolist()
|
| 324 |
+
|
| 325 |
+
|
| 326 |
+
# In[26]:
|
| 327 |
+
|
| 328 |
+
|
| 329 |
+
sc.pl.dotplot(adata,plot_genes,
|
| 330 |
+
"cNMF_cluster", dendrogram=False,standard_scale='var',)
|
| 331 |
+
|
ovrawm/t_commot_flowsig.txt
ADDED
|
@@ -0,0 +1,395 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Spatial Communication
|
| 5 |
+
#
|
| 6 |
+
# Spatial communication is a point of interest for us for the Spatial Transcriptomics Society, and we would like to find the conduction process of spatial communication.
|
| 7 |
+
#
|
| 8 |
+
# Here, we introduce two method integrated in omicverse named `COMMOT` and `flowsig`.
|
| 9 |
+
#
|
| 10 |
+
# We made three improvements in integrating the `COMMOT` and `flowsig` algorithm in OmicVerse:
|
| 11 |
+
#
|
| 12 |
+
# - We reduced the installation conflict of `COMMOT` and `flowsig`, user only need to update OmicVerse to the latest version.
|
| 13 |
+
# - We optimized the visualization of `COMMOT` and `flowsig` and unified the data preprocessing process so that users don't need to struggle with different data processing flows.
|
| 14 |
+
# - We have fixed some bugs that could occur during function.
|
| 15 |
+
#
|
| 16 |
+
# If you found this tutorial helpful, please cite `COMMOT`, `flowsig` and OmicVerse:
|
| 17 |
+
#
|
| 18 |
+
# - Cang, Z., Zhao, Y., Almet, A.A. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat Methods 20, 218–228 (2023). https://doi.org/10.1038/s41592-022-01728-4
|
| 19 |
+
# - Almet, A.A., Tsai, YC., Watanabe, M. et al. Inferring pattern-driving intercellular flows from single-cell and spatial transcriptomics. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02380-w
|
| 20 |
+
|
| 21 |
+
# In[1]:
|
| 22 |
+
|
| 23 |
+
|
| 24 |
+
import omicverse as ov
|
| 25 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 26 |
+
import scanpy as sc
|
| 27 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 28 |
+
ov.plot_set()
|
| 29 |
+
|
| 30 |
+
|
| 31 |
+
# ## Preprocess data
|
| 32 |
+
#
|
| 33 |
+
# Here we present our re-analysis of 151676 sample of the dorsolateral prefrontal cortex (DLPFC) dataset. Maynard et al. has manually annotated DLPFC layers and white matter (WM) based on the morphological features and gene markers.
|
| 34 |
+
#
|
| 35 |
+
# This tutorial demonstrates how to identify spatial domains on 10x Visium data using STAGATE. The processed data are available at https://github.com/LieberInstitute/spatialLIBD. We downloaded the manual annotation from the spatialLIBD package and provided at https://drive.google.com/drive/folders/10lhz5VY7YfvHrtV40MwaqLmWz56U9eBP?usp=sharing.
|
| 36 |
+
|
| 37 |
+
# In[40]:
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
adata = sc.read_visium(path='data', count_file='151676_filtered_feature_bc_matrix.h5')
|
| 41 |
+
adata.var_names_make_unique()
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# <div class="admonition warning">
|
| 45 |
+
# <p class="admonition-title">Note</p>
|
| 46 |
+
# <p>
|
| 47 |
+
# We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code.
|
| 48 |
+
# </p>
|
| 49 |
+
# </div>
|
| 50 |
+
#
|
| 51 |
+
# ```python
|
| 52 |
+
# #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 53 |
+
# #adata.raw = adata
|
| 54 |
+
# #adata = adata[:, adata.var.highly_variable_features]
|
| 55 |
+
# ```
|
| 56 |
+
|
| 57 |
+
# In[ ]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
sc.pp.calculate_qc_metrics(adata, inplace=True)
|
| 61 |
+
adata = adata[:,adata.var['total_counts']>100]
|
| 62 |
+
adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",)
|
| 63 |
+
adata
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
# In[ ]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
adata.write('data/cluster_svg.h5ad',compression='gzip')
|
| 70 |
+
|
| 71 |
+
|
| 72 |
+
# In[3]:
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
#adata=ov.read('data/cluster_svg.h5ad',compression='gzip')
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# ## Communication Analysis with COMMOT
|
| 79 |
+
#
|
| 80 |
+
# ### Spatial communication inference
|
| 81 |
+
#
|
| 82 |
+
# We will use the CellChatDB ligand-receptor database here. Only the secreted signaling LR pairs will be used.
|
| 83 |
+
#
|
| 84 |
+
# Jin, Suoqin, et al. “Inference and analysis of cell-cell communication using CellChat.” Nature communications 12.1 (2021): 1-20.
|
| 85 |
+
|
| 86 |
+
# In[42]:
|
| 87 |
+
|
| 88 |
+
|
| 89 |
+
df_cellchat = ov.externel.commot.pp.ligand_receptor_database(species='human',
|
| 90 |
+
signaling_type='Secreted Signaling',
|
| 91 |
+
database='CellChat')
|
| 92 |
+
print(df_cellchat.shape)
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
# We then filter the LR pairs to keep only the pairs with both ligand and receptor expressed in at least 5% of the spots.
|
| 96 |
+
|
| 97 |
+
# In[43]:
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
df_cellchat_filtered = ov.externel.commot.pp.filter_lr_database(df_cellchat,
|
| 101 |
+
adata,
|
| 102 |
+
min_cell_pct=0.05)
|
| 103 |
+
print(df_cellchat_filtered.shape)
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
# Now perform spatial communication inference for these 250 ligand-receptor pairs with a spatial distance limit of 500. CellChat database considers heteromeric units. The signaling results are stored as spot-by-spot matrices in the obsp slots. For example, the score for spot i signaling to spot j through the LR pair can be retrieved from `adata.obsp['commot-cellchat-Wnt4-Fzd4_Lrp6'][i,j]`.
|
| 107 |
+
|
| 108 |
+
# In[44]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
ov.externel.commot.tl.spatial_communication(adata,
|
| 112 |
+
database_name='cellchat',
|
| 113 |
+
df_ligrec=df_cellchat_filtered,
|
| 114 |
+
dis_thr=500, heteromeric=True,
|
| 115 |
+
pathway_sum=True)
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# (Optional) We read the ground truth area of our spatial data
|
| 119 |
+
#
|
| 120 |
+
# This step is not mandatory to run, in the tutorial, it's just to demonstrate the accuracy of our clustering effect, and in your own tasks, there is often no Ground_truth
|
| 121 |
+
#
|
| 122 |
+
# <div class="admonition warning">
|
| 123 |
+
# <p class="admonition-title">Note</p>
|
| 124 |
+
# <p>
|
| 125 |
+
# You can also use Celltype and other annotated results in adata.obs, here is just a randomly selected type, there is no particular significance, in order to facilitate the visualization and study the signal
|
| 126 |
+
# </p>
|
| 127 |
+
# </div>
|
| 128 |
+
|
| 129 |
+
# In[45]:
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# read the annotation
|
| 133 |
+
import pandas as pd
|
| 134 |
+
import os
|
| 135 |
+
Ann_df = pd.read_csv(os.path.join('data', '151676_truth.txt'), sep='\t', header=None, index_col=0)
|
| 136 |
+
Ann_df.columns = ['Ground_Truth']
|
| 137 |
+
adata.obs['Ground_Truth'] = Ann_df.loc[adata.obs_names, 'Ground_Truth']
|
| 138 |
+
Layer_color=['#283b5c', '#d8e17b', '#838e44', '#4e8991', '#d08c35', '#511a3a',
|
| 139 |
+
'#c2c2c2', '#dfc648']
|
| 140 |
+
sc.pl.spatial(adata, img_key="hires", color=["Ground_Truth"],palette=Layer_color)
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
# ### Visualize the communication signal in spatial space
|
| 144 |
+
#
|
| 145 |
+
# Determine the spatial direction of a signaling pathway, for example, the FGF pathway. The interpolated signaling directions for where the signals are sent by the spots and where the signals received by the spots are from are stored in `adata.obsm['commot_sender_vf-cellchat-FGF']` and `adata.obsm['commot_receiver_vf-cellchat-FGF']`, respectively.
|
| 146 |
+
#
|
| 147 |
+
# Taken together, our findings indicate that FGF signaling is crucial for cortical folding in gyrencephalic mammals and is a pivotal upstream regulator of the production of OSVZ progenitors. FGF signaling is the first signaling pathway found to regulate cortical folding.
|
| 148 |
+
|
| 149 |
+
# In[46]:
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
ct_color_dict=dict(zip(adata.obs['Ground_Truth'].cat.categories,
|
| 153 |
+
adata.uns['Ground_Truth_colors']))
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# In[47]:
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
adata.uns['commot-cellchat-info']['df_ligrec'].head()
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
# In[48]:
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
import matplotlib.pyplot as plt
|
| 166 |
+
scale=0.000008
|
| 167 |
+
k=5
|
| 168 |
+
goal_pathway='FGF'
|
| 169 |
+
ov.externel.commot.tl.communication_direction(adata, database_name='cellchat', pathway_name=goal_pathway, k=k)
|
| 170 |
+
ov.externel.commot.pl.plot_cell_communication(adata, database_name='cellchat',
|
| 171 |
+
pathway_name='FGF', plot_method='grid',
|
| 172 |
+
background_legend=True,
|
| 173 |
+
scale=scale, ndsize=8, grid_density=0.4,
|
| 174 |
+
summary='sender', background='cluster',
|
| 175 |
+
clustering='Ground_Truth',
|
| 176 |
+
cluster_cmap=ct_color_dict,
|
| 177 |
+
cmap='Alphabet',
|
| 178 |
+
normalize_v = True, normalize_v_quantile=0.995)
|
| 179 |
+
plt.title(f'Pathway:{goal_pathway}',fontsize=13)
|
| 180 |
+
#plt.savefig('figures/TLE/TLE_cellchat_all_FGF.png',dpi=300,bbox_inches='tight')
|
| 181 |
+
#fig.savefig('pdf/TLE/control_cellchat_all_FGF.pdf',dpi=300,bbox_inches='tight')
|
| 182 |
+
|
| 183 |
+
|
| 184 |
+
# In[49]:
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
adata.write('data/151676_commot.h5ad',compression='gzip')
|
| 188 |
+
|
| 189 |
+
|
| 190 |
+
# In[2]:
|
| 191 |
+
|
| 192 |
+
|
| 193 |
+
adata=ov.read('data/151676_commot.h5ad')
|
| 194 |
+
adata
|
| 195 |
+
|
| 196 |
+
|
| 197 |
+
# ## Communication signal inference with flowsig
|
| 198 |
+
#
|
| 199 |
+
# ### Construct GEMs
|
| 200 |
+
# We now construct gene expression modules (GEMs) from the unnormalised count data. For ST data, we use `NMF`.
|
| 201 |
+
|
| 202 |
+
# In[3]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
adata.layers['normalized'] = adata.X.copy()
|
| 206 |
+
|
| 207 |
+
# We construct 10 gene expression modules using the raw cell count.
|
| 208 |
+
ov.externel.flowsig.pp.construct_gems_using_nmf(adata,
|
| 209 |
+
n_gems = 10,
|
| 210 |
+
layer_key = 'counts',
|
| 211 |
+
)
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
# If you want to study the genes in a GEM, we provide the `ov.externel.flowsig.ul.get_top_gem_genes` function for getting the genes in a specific GEM.
|
| 215 |
+
|
| 216 |
+
# In[4]:
|
| 217 |
+
|
| 218 |
+
|
| 219 |
+
goal_gem='GEM-5'
|
| 220 |
+
gem_gene=ov.externel.flowsig.ul.get_top_gem_genes(adata=adata,
|
| 221 |
+
gems=[goal_gem],
|
| 222 |
+
n_genes=100,
|
| 223 |
+
gene_type='all',
|
| 224 |
+
method = 'nmf',
|
| 225 |
+
)
|
| 226 |
+
gem_gene.head()
|
| 227 |
+
|
| 228 |
+
|
| 229 |
+
# ### Construct the flow expression matrices
|
| 230 |
+
#
|
| 231 |
+
# We construct augmented flow expression matrices for each condition that measure three types of variables:
|
| 232 |
+
#
|
| 233 |
+
# 1. Intercellular signal inflow, i.e., how much of a signal did a cell receive. For ST data, signal inflow is constructed by summing the received signals for each significant ligand inferred by COMMOT.
|
| 234 |
+
# 2. GEMs, which encapsulate intracellular information processing. We define these as cellwise membership to the GEM.
|
| 235 |
+
# Intercellular signal outflow, i.e., how much of a signal did a cell send. These are simply ligand gene expression.
|
| 236 |
+
# 3. The kay assumption of flowsig is that all intercellular information flows are directed from signal inflows to GEMs, from one GEM to another GEM, and from GEMs to signal outflows.
|
| 237 |
+
#
|
| 238 |
+
# For spatial data, we use COMMOT output directly to construct signal inflow expression and do not need knowledge about TF databases.
|
| 239 |
+
|
| 240 |
+
# In[5]:
|
| 241 |
+
|
| 242 |
+
|
| 243 |
+
commot_output_key = 'commot-cellchat'
|
| 244 |
+
# We first construct the potential cellular flows from the commot output
|
| 245 |
+
ov.externel.flowsig.pp.construct_flows_from_commot(adata,
|
| 246 |
+
commot_output_key,
|
| 247 |
+
gem_expr_key = 'X_gem',
|
| 248 |
+
scale_gem_expr = True,
|
| 249 |
+
flowsig_network_key = 'flowsig_network',
|
| 250 |
+
flowsig_expr_key = 'X_flow')
|
| 251 |
+
|
| 252 |
+
|
| 253 |
+
# For spatial data, we retain spatially informative variables, which we determine by calculating the Moran's I value for signal inflow and signal outflow variables. In case the spatial graph has not been calculated for this data yet, FlowSig will do so, meaning that we need to specify both the coordinate type, grid or generic, and in the case of the former, n_neighs, which in this case, is 8.
|
| 254 |
+
#
|
| 255 |
+
# Flow expression variables are defined to be spatially informative if their Moran's I value is above a specified threshold.
|
| 256 |
+
|
| 257 |
+
# In[6]:
|
| 258 |
+
|
| 259 |
+
|
| 260 |
+
# Then we subset for "spatially flowing" inflows and outflows
|
| 261 |
+
ov.externel.flowsig.pp.determine_informative_variables(adata,
|
| 262 |
+
flowsig_expr_key = 'X_flow',
|
| 263 |
+
flowsig_network_key = 'flowsig_network',
|
| 264 |
+
spatial = True,
|
| 265 |
+
moran_threshold = 0.15,
|
| 266 |
+
coord_type = 'grid',
|
| 267 |
+
n_neighbours = 8,
|
| 268 |
+
library_key = None)
|
| 269 |
+
|
| 270 |
+
|
| 271 |
+
# ### Learn intercellular flows
|
| 272 |
+
#
|
| 273 |
+
# For spatial data, where there are far fewer "control vs. perturbed" studies, we use the GSP method, which uses conditional independence testing and a greedy algorithm to learn the CPDAG containing directed arcs and undirected edges.
|
| 274 |
+
#
|
| 275 |
+
# For spatial data, we cannot bootstrap by resampling across individual cells because we would lose the additional layer of correlation contained in the spatial data. Rather, we divide the tissue up into spatial "blocks" and resample within blocks. This is known as block bootstrapping.
|
| 276 |
+
#
|
| 277 |
+
# To calculate the blocks, we used scikit-learn's k-means clustering method to generate 20 roughly equally sized spatial blocks.
|
| 278 |
+
|
| 279 |
+
# In[9]:
|
| 280 |
+
|
| 281 |
+
|
| 282 |
+
from sklearn.cluster import KMeans
|
| 283 |
+
import pandas as pd
|
| 284 |
+
|
| 285 |
+
kmeans = KMeans(n_clusters=10, random_state=0).fit(adata.obsm['spatial'])
|
| 286 |
+
adata.obs['spatial_kmeans'] = pd.Series(kmeans.labels_, dtype='category').values
|
| 287 |
+
|
| 288 |
+
|
| 289 |
+
# We use these blocks to learn the spatial intercellular flows.
|
| 290 |
+
|
| 291 |
+
# In[ ]:
|
| 292 |
+
|
| 293 |
+
|
| 294 |
+
# # Now we are ready to learn the network
|
| 295 |
+
ov.externel.flowsig.tl.learn_intercellular_flows(adata,
|
| 296 |
+
flowsig_key = 'flowsig_network',
|
| 297 |
+
flow_expr_key = 'X_flow',
|
| 298 |
+
use_spatial = True,
|
| 299 |
+
block_key = 'spatial_kmeans',
|
| 300 |
+
n_jobs = 4,
|
| 301 |
+
n_bootstraps = 500)
|
| 302 |
+
|
| 303 |
+
|
| 304 |
+
# ### Partially validate intercellular flow network
|
| 305 |
+
#
|
| 306 |
+
# Finally, we will remove any "false positive" edges. Noting that the CPDAG contains directed arcs and undirected arcs we do two things.
|
| 307 |
+
#
|
| 308 |
+
# First, we remove directed arcs that are not oriented from signal inflow to GEM, GEM to GEM, or from GEM to signal outflow and for undirected edges, we reorient them so that they obey the previous directionalities.
|
| 309 |
+
|
| 310 |
+
# In[8]:
|
| 311 |
+
|
| 312 |
+
|
| 313 |
+
# This part is key for reducing false positives
|
| 314 |
+
ov.externel.flowsig.tl.apply_biological_flow(adata,
|
| 315 |
+
flowsig_network_key = 'flowsig_network',
|
| 316 |
+
adjacency_key = 'adjacency',
|
| 317 |
+
validated_key = 'validated')
|
| 318 |
+
|
| 319 |
+
|
| 320 |
+
# Second, we will remove directed arcs whose bootstrapped frequencies are below a specified edge threshold as well as undirected edges whose total bootstrapped frequencies are below the same threshold. Because we did not have perturbation data, we specify a more stringent edge threshold.
|
| 321 |
+
#
|
| 322 |
+
|
| 323 |
+
# In[26]:
|
| 324 |
+
|
| 325 |
+
|
| 326 |
+
edge_threshold = 0.7
|
| 327 |
+
|
| 328 |
+
ov.externel.flowsig.tl.filter_low_confidence_edges(adata,
|
| 329 |
+
edge_threshold = edge_threshold,
|
| 330 |
+
flowsig_network_key = 'flowsig_network',
|
| 331 |
+
adjacency_key = 'adjacency_validated',
|
| 332 |
+
filtered_key = 'filtered')
|
| 333 |
+
|
| 334 |
+
|
| 335 |
+
# In[27]:
|
| 336 |
+
|
| 337 |
+
|
| 338 |
+
adata.write('data/cortex_commot_flowsig.h5ad',compression='gzip')
|
| 339 |
+
|
| 340 |
+
|
| 341 |
+
# In[2]:
|
| 342 |
+
|
| 343 |
+
|
| 344 |
+
#adata=ov.read('data/cortex_commot_flowsig.h5ad')
|
| 345 |
+
|
| 346 |
+
|
| 347 |
+
# ## Visualize the result of flowsig
|
| 348 |
+
#
|
| 349 |
+
# We can construct the directed NetworkX DiGraph object from adjacency_validated_filtered.
|
| 350 |
+
|
| 351 |
+
# In[3]:
|
| 352 |
+
|
| 353 |
+
|
| 354 |
+
flow_network = ov.externel.flowsig.tl.construct_intercellular_flow_network(adata,
|
| 355 |
+
flowsig_network_key = 'flowsig_network',
|
| 356 |
+
adjacency_key = 'adjacency_validated_filtered')
|
| 357 |
+
|
| 358 |
+
|
| 359 |
+
# ### Cell-specific GEM
|
| 360 |
+
#
|
| 361 |
+
# The first thing we need to be concerned about is which GEM, exactly, is relevant to the cell I want to study. Here, we use dotplot to visualize the expression of GEM in different cell types.
|
| 362 |
+
|
| 363 |
+
# In[4]:
|
| 364 |
+
|
| 365 |
+
|
| 366 |
+
flowsig_expr_key='X_gem'
|
| 367 |
+
X_flow = adata.obsm[flowsig_expr_key]
|
| 368 |
+
adata_subset = sc.AnnData(X=X_flow)
|
| 369 |
+
adata_subset.obs = adata.obs
|
| 370 |
+
adata_subset.var.index =[f'GEM-{i}' for i in range(1,len(adata_subset.var)+1)]
|
| 371 |
+
|
| 372 |
+
|
| 373 |
+
# In[5]:
|
| 374 |
+
|
| 375 |
+
|
| 376 |
+
import matplotlib.pyplot as plt
|
| 377 |
+
ax=sc.pl.dotplot(adata_subset, adata_subset.var.index, groupby='Ground_Truth',
|
| 378 |
+
dendrogram=True,standard_scale='var',cmap='Reds',show=False)
|
| 379 |
+
color_dict=dict(zip(adata.obs['Ground_Truth'].cat.categories,adata.uns['Ground_Truth_colors']))
|
| 380 |
+
|
| 381 |
+
|
| 382 |
+
# ### Visualize the flowsig network
|
| 383 |
+
#
|
| 384 |
+
# We fixed the network function provided by the author and provided a better visualization.
|
| 385 |
+
|
| 386 |
+
# In[7]:
|
| 387 |
+
|
| 388 |
+
|
| 389 |
+
ov.pl.plot_flowsig_network(flow_network=flow_network,
|
| 390 |
+
gem_plot=['GEM-2','GEM-7','GEM-1','GEM-3','GEM-4','GEM-5'],
|
| 391 |
+
figsize=(8,4),
|
| 392 |
+
curve_awarg={'eps':2},
|
| 393 |
+
node_shape={'GEM':'^','Sender':'o','Receptor':'o'},
|
| 394 |
+
ylim=(-0.5,0.5),xlim=(-3,3))
|
| 395 |
+
|
ovrawm/t_cytotrace.txt
ADDED
|
@@ -0,0 +1,110 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Prediction of absolute developmental potential using CytoTrace2
|
| 5 |
+
#
|
| 6 |
+
# CytoTRACE 2 is a computational method for predicting cellular potency categories and absolute developmental potential from single-cell RNA-sequencing data.
|
| 7 |
+
#
|
| 8 |
+
# Potency categories in the context of CytoTRACE 2 classify cells based on their developmental potential, ranging from totipotent and pluripotent cells with broad differentiation potential to lineage-restricted oligopotent, multipotent and unipotent cells capable of producing varying numbers of downstream cell types, and finally, differentiated cells, ranging from mature to terminally differentiated phenotypes.
|
| 9 |
+
#
|
| 10 |
+
# We made three improvements in integrating the CytoTrace2 algorithm in OmicVerse:
|
| 11 |
+
#
|
| 12 |
+
# - No additional packages to install, including R
|
| 13 |
+
# - We fixed a bug in multi-threaded pools to avoid potential error reporting
|
| 14 |
+
# - Native support for `anndata`, you don't need to export `input_file` and `annotation_file`.
|
| 15 |
+
#
|
| 16 |
+
# If you found this tutorial helpful, please cite CytoTrace2 and OmicVerse:
|
| 17 |
+
#
|
| 18 |
+
# Kang, M., Armenteros, J. J. A., Gulati, G. S., Gleyzer, R., Avagyan, S., Brown, E. L., Zhang, W., Usmani, A., Earland, N., Wu, Z., Zou, J., Fields, R. C., Chen, D. Y., Chaudhuri, A. A., & Newman, A. M. (2024). Mapping single-cell developmental potential in health and disease with interpretable deep learning. bioRxiv : the preprint server for biology, 2024.03.19.585637. https://doi.org/10.1101/2024.03.19.585637
|
| 19 |
+
|
| 20 |
+
# In[1]:
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
import omicverse as ov
|
| 24 |
+
ov.plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# ## Preprocess data
|
| 28 |
+
#
|
| 29 |
+
# As an example, we apply differential kinetic analysis to dentate gyrus neurogenesis, which comprises multiple heterogeneous subpopulations.
|
| 30 |
+
|
| 31 |
+
# In[2]:
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
import scvelo as scv
|
| 35 |
+
adata=scv.datasets.dentategyrus()
|
| 36 |
+
adata
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
# In[4]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)\nadata\n")
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# ## Predict cytotrace2
|
| 46 |
+
#
|
| 47 |
+
# We need to import the two pre-trained models from CytoTrace2, see the download links for the models:
|
| 48 |
+
#
|
| 49 |
+
# - Figshare:
|
| 50 |
+
# https://figshare.com/ndownloader/files/47258749
|
| 51 |
+
#
|
| 52 |
+
# - or Github:
|
| 53 |
+
# https://github.com/digitalcytometry/cytotrace2/tree/main/cytotrace2_python/cytotrace2_py/resources/17_models_weights
|
| 54 |
+
# https://github.com/digitalcytometry/cytotrace2/tree/main/cytotrace2_python/cytotrace2_py/resources/5_models_weights
|
| 55 |
+
#
|
| 56 |
+
# All parameters are explained as follows:
|
| 57 |
+
# - adata: AnnData object containing the scRNA-seq data.
|
| 58 |
+
# - use_model_dir: Path to the directory containing the pre-trained model files.
|
| 59 |
+
# - species: The species of the input data. Default is "mouse".
|
| 60 |
+
# - batch_size: The number of cells to process in each batch. Default is 10000.
|
| 61 |
+
# - smooth_batch_size: The number of cells to process in each batch for smoothing. Default is 1000.
|
| 62 |
+
# - disable_parallelization: If True, disable parallel processing. Default is False.
|
| 63 |
+
# - max_cores: Maximum number of CPU cores to use for parallel processing. If None, all available cores will be used. Default is None.
|
| 64 |
+
# - max_pcs: Maximum number of principal components to use. Default is 200.
|
| 65 |
+
# - seed: Random seed for reproducibility. Default is 14.
|
| 66 |
+
# - output_dir: Directory to save the results. Default is 'cytotrace2_results'.
|
| 67 |
+
|
| 68 |
+
# In[5]:
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
results = ov.single.cytotrace2(adata,
|
| 72 |
+
use_model_dir="cymodels/5_models_weights",
|
| 73 |
+
species="mouse",
|
| 74 |
+
batch_size = 10000,
|
| 75 |
+
smooth_batch_size = 1000,
|
| 76 |
+
disable_parallelization = False,
|
| 77 |
+
max_cores = None,
|
| 78 |
+
max_pcs = 200,
|
| 79 |
+
seed = 14,
|
| 80 |
+
output_dir = 'cytotrace2_results'
|
| 81 |
+
)
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
# ## Visualizing
|
| 85 |
+
#
|
| 86 |
+
# Visualizing the results we can directly compare the predicted potency scores with the known developmental stage of the cells, seeing how the predictions meticulously align with the known biology. Take a look!
|
| 87 |
+
|
| 88 |
+
# In[8]:
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 92 |
+
color=['clusters','CytoTRACE2_Score'],
|
| 93 |
+
frameon='small',cmap='Reds',wspace=0.55)
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
# - Left: demonstrates the distribution of different cell types in UMAP space.
|
| 97 |
+
# - Right: demonstrates the CytoTRACE 2 scores of different cell types; cells with high scores are generally considered to have a higher pluripotency or undifferentiated state.
|
| 98 |
+
|
| 99 |
+
# In[9]:
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 103 |
+
color=['CytoTRACE2_Potency','CytoTRACE2_Relative'],
|
| 104 |
+
frameon='small',cmap='Reds',wspace=0.55)
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
# - Potency category:
|
| 108 |
+
# The UMAP embedding plot of predicted potency category reflects the discrete classification of cells into potency categories, taking possible values of Differentiated, Unipotent, Oligopotent, Multipotent, Pluripotent, and Totipotent.
|
| 109 |
+
# - Relative order:
|
| 110 |
+
# UMAP embedding of predicted relative order, which is based on absolute predicted potency scores normalized to the range 0 (more differentiated) to 1 (less differentiated). Provides the relative ordering of cells by developmental potential
|
ovrawm/t_deg.txt
ADDED
|
@@ -0,0 +1,323 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Different Expression Analysis
|
| 5 |
+
#
|
| 6 |
+
# An important task of bulk rna-seq analysis is the different expression , which we can perform with omicverse. For different expression analysis, ov change the `gene_id` to `gene_name` of matrix first. When our dataset existed the batch effect, we can use the SizeFactors of DEseq2 to normalize it, and use `t-test` of `wilcoxon` to calculate the p-value of genes. Here we demonstrate this pipeline with a matrix from `featureCounts`. The same pipeline would generally be used to analyze any collection of RNA-seq tasks.
|
| 7 |
+
#
|
| 8 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1q5lDfJepbtvNtc1TKz-h4wGUifTZ3i0_?usp=sharing
|
| 9 |
+
|
| 10 |
+
# In[1]:
|
| 11 |
+
|
| 12 |
+
|
| 13 |
+
import omicverse as ov
|
| 14 |
+
import scanpy as sc
|
| 15 |
+
import matplotlib.pyplot as plt
|
| 16 |
+
|
| 17 |
+
ov.plot_set()
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
# ## Geneset Download
|
| 21 |
+
#
|
| 22 |
+
# When we need to convert a gene id, we need to prepare a mapping pair file. Here we have pre-processed 6 genome gtf files and generated mapping pairs including `T2T-CHM13`, `GRCh38`, `GRCh37`, `GRCm39`, `danRer7`, and `danRer11`. If you need to convert other id_mapping, you can generate your own mapping using gtf Place the files in the `genesets` directory.
|
| 23 |
+
|
| 24 |
+
# In[2]:
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
ov.utils.download_geneid_annotation_pair()
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
# Note that this dataset has not been processed in any way and is only exported by `featureCounts`, and Sequence alignment was performed from the genome file of CRCm39
|
| 31 |
+
#
|
| 32 |
+
# sample data can be download from: https://raw.githubusercontent.com/Starlitnightly/omicverse/master/sample/counts.txt
|
| 33 |
+
|
| 34 |
+
# In[3]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
#data=pd.read_csv('https://raw.githubusercontent.com/Starlitnightly/omicverse/master/sample/counts.txt',index_col=0,sep='\t',header=1)
|
| 38 |
+
data=ov.read('data/counts.txt',index_col=0,header=1)
|
| 39 |
+
#replace the columns `.bam` to ``
|
| 40 |
+
data.columns=[i.split('/')[-1].replace('.bam','') for i in data.columns]
|
| 41 |
+
data.head()
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# ## ID mapping
|
| 45 |
+
#
|
| 46 |
+
# We performed the gene_id mapping by the mapping pair file `GRCm39` downloaded before.
|
| 47 |
+
|
| 48 |
+
# In[4]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
data=ov.bulk.Matrix_ID_mapping(data,'genesets/pair_GRCm39.tsv')
|
| 52 |
+
data.head()
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
# ## Different expression analysis with ov
|
| 56 |
+
#
|
| 57 |
+
# We can do differential expression analysis very simply by ov, simply by providing an expression matrix. To run DEG, we simply need to:
|
| 58 |
+
#
|
| 59 |
+
# - Read the raw count by featureCount or any other qualify methods.
|
| 60 |
+
# - Create an ov DEseq object.
|
| 61 |
+
|
| 62 |
+
# In[5]:
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
dds=ov.bulk.pyDEG(data)
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
# We notes that the gene_name mapping before exist some duplicates, we will process the duplicate indexes to retain only the highest expressed genes
|
| 69 |
+
|
| 70 |
+
# In[6]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
dds.drop_duplicates_index()
|
| 74 |
+
print('... drop_duplicates_index success')
|
| 75 |
+
|
| 76 |
+
|
| 77 |
+
# We also need to remove the batch effect of the expression matrix, `estimateSizeFactors` of DEseq2 to be used to normalize our matrix
|
| 78 |
+
|
| 79 |
+
# In[7]:
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
dds.normalize()
|
| 83 |
+
print('... estimateSizeFactors and normalize success')
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# Now we can calculate the different expression gene from matrix, we need to input the treatment and control groups
|
| 87 |
+
|
| 88 |
+
# In[8]:
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
treatment_groups=['4-3','4-4']
|
| 92 |
+
control_groups=['1--1','1--2']
|
| 93 |
+
result=dds.deg_analysis(treatment_groups,control_groups,method='ttest')
|
| 94 |
+
result.head()
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# One important thing is that we do not filter out low expression genes when processing DEGs, and in future versions I will consider building in the corresponding processing.
|
| 98 |
+
|
| 99 |
+
# In[9]:
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
print(result.shape)
|
| 103 |
+
result=result.loc[result['log2(BaseMean)']>1]
|
| 104 |
+
print(result.shape)
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
# We also need to set the threshold of Foldchange, we prepare a method named `foldchange_set` to finish. This function automatically calculates the appropriate threshold based on the log2FC distribution, but you can also enter it manually.
|
| 108 |
+
|
| 109 |
+
# In[10]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
# -1 means automatically calculates
|
| 113 |
+
dds.foldchange_set(fc_threshold=-1,
|
| 114 |
+
pval_threshold=0.05,
|
| 115 |
+
logp_max=6)
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# ## Visualize the DEG result and specific genes
|
| 119 |
+
#
|
| 120 |
+
# To visualize the DEG result, we use `plot_volcano` to do it. This fuction can visualize the gene interested or high different expression genes. There are some parameters you need to input:
|
| 121 |
+
#
|
| 122 |
+
# - title: The title of volcano
|
| 123 |
+
# - figsize: The size of figure
|
| 124 |
+
# - plot_genes: The genes you interested
|
| 125 |
+
# - plot_genes_num: If you don't have interested genes, you can auto plot it.
|
| 126 |
+
|
| 127 |
+
# In[11]:
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
|
| 131 |
+
plot_genes_num=8,plot_genes_fontsize=12,)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# To visualize the specific genes, we only need to use the `dds.plot_boxplot` function to finish it.
|
| 135 |
+
|
| 136 |
+
# In[12]:
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
dds.plot_boxplot(genes=['Ckap2','Lef1'],treatment_groups=treatment_groups,
|
| 140 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 141 |
+
legend_bbox=(2,0.55))
|
| 142 |
+
|
| 143 |
+
|
| 144 |
+
# In[13]:
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
dds.plot_boxplot(genes=['Ckap2'],treatment_groups=treatment_groups,
|
| 148 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 149 |
+
legend_bbox=(2,0.55))
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
# ## Pathway enrichment analysis by ov
|
| 153 |
+
#
|
| 154 |
+
# Here we use the `gseapy` package, which included the GSEA analysis and Enrichment. We have optimised the output of the package and given some better looking graph drawing functions
|
| 155 |
+
#
|
| 156 |
+
# Similarly, we need to download the pathway/genesets first. Five genesets we prepare previously, you can use `ov.utils.download_pathway_database()` to download automatically. Besides, you can download the pathway you interested from enrichr: https://maayanlab.cloud/Enrichr/#libraries
|
| 157 |
+
|
| 158 |
+
# In[37]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
ov.utils.download_pathway_database()
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
# In[14]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/WikiPathways_2019_Mouse.txt',organism='Mouse')
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
# Note that the `pvalue_type` we set to `auto`, this is because when the genesets we enrichment if too small, use the `adjusted pvalue` we can't get the correct result. So you can set `adjust` or `raw` to get the significant geneset.
|
| 171 |
+
#
|
| 172 |
+
# If you didn't have internet, please set `background` to all genes expressed in rna-seq,like:
|
| 173 |
+
#
|
| 174 |
+
# ```python
|
| 175 |
+
# enr=ov.bulk.geneset_enrichment(gene_list=deg_genes,
|
| 176 |
+
# pathways_dict=pathway_dict,
|
| 177 |
+
# pvalue_type='auto',
|
| 178 |
+
# background=dds.result.index.tolist(),
|
| 179 |
+
# organism='mouse')
|
| 180 |
+
# ```
|
| 181 |
+
|
| 182 |
+
# In[15]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
deg_genes=dds.result.loc[dds.result['sig']!='normal'].index.tolist()
|
| 186 |
+
enr=ov.bulk.geneset_enrichment(gene_list=deg_genes,
|
| 187 |
+
pathways_dict=pathway_dict,
|
| 188 |
+
pvalue_type='auto',
|
| 189 |
+
organism='mouse')
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
# To visualize the enrichment, we use `geneset_plot` to finish it
|
| 193 |
+
|
| 194 |
+
# In[21]:
|
| 195 |
+
|
| 196 |
+
|
| 197 |
+
ov.bulk.geneset_plot(enr,figsize=(2,5),fig_title='Wiki Pathway enrichment',
|
| 198 |
+
cax_loc=[2, 0.45, 0.5, 0.02],
|
| 199 |
+
bbox_to_anchor_used=(-0.25, -13),node_diameter=10,
|
| 200 |
+
custom_ticks=[5,7],text_knock=3,
|
| 201 |
+
cmap='Reds')
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
# ## Multi pathway enrichment
|
| 205 |
+
#
|
| 206 |
+
# In addition to pathway enrichment for a single database, OmicVerse supports enriching and visualizing multiple pathways at the same time, which is implemented using [`pyComplexHeatmap`](https://dingwb.github.io/PyComplexHeatmap/build/html/notebooks/gene_enrichment_analysis.html), and Citetation is welcome!
|
| 207 |
+
|
| 208 |
+
# In[22]:
|
| 209 |
+
|
| 210 |
+
|
| 211 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Biological_Process_2023.txt',organism='Mouse')
|
| 212 |
+
enr_go_bp=ov.bulk.geneset_enrichment(gene_list=deg_genes,
|
| 213 |
+
pathways_dict=pathway_dict,
|
| 214 |
+
pvalue_type='auto',
|
| 215 |
+
organism='mouse')
|
| 216 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Molecular_Function_2023.txt',organism='Mouse')
|
| 217 |
+
enr_go_mf=ov.bulk.geneset_enrichment(gene_list=deg_genes,
|
| 218 |
+
pathways_dict=pathway_dict,
|
| 219 |
+
pvalue_type='auto',
|
| 220 |
+
organism='mouse')
|
| 221 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/GO_Cellular_Component_2023.txt',organism='Mouse')
|
| 222 |
+
enr_go_cc=ov.bulk.geneset_enrichment(gene_list=deg_genes,
|
| 223 |
+
pathways_dict=pathway_dict,
|
| 224 |
+
pvalue_type='auto',
|
| 225 |
+
organism='mouse')
|
| 226 |
+
|
| 227 |
+
|
| 228 |
+
# In[167]:
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
enr_dict={'BP':enr_go_bp,
|
| 232 |
+
'MF':enr_go_mf,
|
| 233 |
+
'CC':enr_go_cc}
|
| 234 |
+
colors_dict={
|
| 235 |
+
'BP':ov.pl.red_color[1],
|
| 236 |
+
'MF':ov.pl.green_color[1],
|
| 237 |
+
'CC':ov.pl.blue_color[1],
|
| 238 |
+
}
|
| 239 |
+
|
| 240 |
+
ov.bulk.geneset_plot_multi(enr_dict,colors_dict,num=3,
|
| 241 |
+
figsize=(2,5),
|
| 242 |
+
text_knock=3,fontsize=8,
|
| 243 |
+
cmap='Reds'
|
| 244 |
+
)
|
| 245 |
+
|
| 246 |
+
|
| 247 |
+
# In[166]:
|
| 248 |
+
|
| 249 |
+
|
| 250 |
+
def geneset_plot_multi(enr_dict,colors_dict,num:int=5,fontsize=10,
|
| 251 |
+
fig_title:str='',fig_xlabel:str='Fractions of genes',
|
| 252 |
+
figsize:tuple=(2,4),cmap:str='YlGnBu',
|
| 253 |
+
text_knock:int=5,text_maxsize:int=20,ax=None,
|
| 254 |
+
):
|
| 255 |
+
from PyComplexHeatmap import HeatmapAnnotation,DotClustermapPlotter,anno_label,anno_simple,AnnotationBase
|
| 256 |
+
for key in enr_dict.keys():
|
| 257 |
+
enr_dict[key]['Type']=key
|
| 258 |
+
enr_all=pd.concat([enr_dict[i].iloc[:num] for i in enr_dict.keys()],axis=0)
|
| 259 |
+
enr_all['Term']=[ov.utils.plot_text_set(i.split('(')[0],text_knock=text_knock,text_maxsize=text_maxsize) for i in enr_all.Term.tolist()]
|
| 260 |
+
enr_all.index=enr_all.Term
|
| 261 |
+
enr_all['Term1']=[i for i in enr_all.index.tolist()]
|
| 262 |
+
del enr_all['Term']
|
| 263 |
+
|
| 264 |
+
colors=colors_dict
|
| 265 |
+
|
| 266 |
+
left_ha = HeatmapAnnotation(
|
| 267 |
+
label=anno_label(enr_all.Type, merge=True,rotation=0,colors=colors,relpos=(1,0.8)),
|
| 268 |
+
Category=anno_simple(enr_all.Type,cmap='Set1',
|
| 269 |
+
add_text=False,legend=False,colors=colors),
|
| 270 |
+
axis=0,verbose=0,label_kws={'rotation':45,'horizontalalignment':'left','visible':False})
|
| 271 |
+
right_ha = HeatmapAnnotation(
|
| 272 |
+
label=anno_label(enr_all.Term1, merge=True,rotation=0,relpos=(0,0.5),arrowprops=dict(visible=True),
|
| 273 |
+
colors=enr_all.assign(color=enr_all.Type.map(colors)).set_index('Term1').color.to_dict(),
|
| 274 |
+
fontsize=fontsize,luminance=0.8,height=2),
|
| 275 |
+
axis=0,verbose=0,#label_kws={'rotation':45,'horizontalalignment':'left'},
|
| 276 |
+
orientation='right')
|
| 277 |
+
if ax==None:
|
| 278 |
+
fig, ax = plt.subplots(figsize=figsize)
|
| 279 |
+
else:
|
| 280 |
+
ax=ax
|
| 281 |
+
#plt.figure(figsize=figsize)
|
| 282 |
+
cm = DotClustermapPlotter(data=enr_all, x='fraction',y='Term1',value='logp',c='logp',s='num',
|
| 283 |
+
cmap=cmap,
|
| 284 |
+
row_cluster=True,#col_cluster=True,#hue='Group',
|
| 285 |
+
#cmap={'Group1':'Greens','Group2':'OrRd'},
|
| 286 |
+
vmin=-1*np.log10(0.1),vmax=-1*np.log10(1e-10),
|
| 287 |
+
#colors={'Group1':'yellowgreen','Group2':'orange'},
|
| 288 |
+
#marker={'Group1':'*','Group2':'$\\ast$'},
|
| 289 |
+
show_rownames=True,show_colnames=False,row_dendrogram=False,
|
| 290 |
+
col_names_side='top',row_names_side='right',
|
| 291 |
+
xticklabels_kws={'labelrotation': 30, 'labelcolor': 'blue','labelsize':fontsize},
|
| 292 |
+
#yticklabels_kws={'labelsize':10},
|
| 293 |
+
#top_annotation=col_ha,left_annotation=left_ha,right_annotation=right_ha,
|
| 294 |
+
left_annotation=left_ha,right_annotation=right_ha,
|
| 295 |
+
spines=False,
|
| 296 |
+
row_split=enr_all.Type,# row_split_gap=1,
|
| 297 |
+
#col_split=df_col.Group,col_split_gap=0.5,
|
| 298 |
+
verbose=1,legend_gap=10,
|
| 299 |
+
#dot_legend_marker='*',
|
| 300 |
+
|
| 301 |
+
xlabel='Fractions of genes',xlabel_side="bottom",
|
| 302 |
+
xlabel_kws=dict(labelpad=8,fontweight='normal',fontsize=fontsize+2),
|
| 303 |
+
# xlabel_bbox_kws=dict(facecolor=facecolor)
|
| 304 |
+
)
|
| 305 |
+
tesr=plt.gcf().axes
|
| 306 |
+
for ax in plt.gcf().axes:
|
| 307 |
+
if hasattr(ax, 'get_xlabel'):
|
| 308 |
+
if ax.get_xlabel() == 'Fractions of genes': # 假设 colorbar 有一个特定的标签
|
| 309 |
+
cbar = ax
|
| 310 |
+
cbar.grid(False)
|
| 311 |
+
if ax.get_ylabel() == 'logp': # 假设 colorbar 有一个特定的标签
|
| 312 |
+
cbar = ax
|
| 313 |
+
cbar.tick_params(labelsize=fontsize+2)
|
| 314 |
+
cbar.set_ylabel(r'$−Log_{10}(P_{adjusted})$',fontsize=fontsize+2)
|
| 315 |
+
cbar.grid(False)
|
| 316 |
+
return ax
|
| 317 |
+
|
| 318 |
+
|
| 319 |
+
# In[ ]:
|
| 320 |
+
|
| 321 |
+
|
| 322 |
+
|
| 323 |
+
|
ovrawm/t_deseq2.txt
ADDED
|
@@ -0,0 +1,237 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Different Expression Analysis with DEseq2
|
| 5 |
+
#
|
| 6 |
+
# An important task of bulk rna-seq analysis is the different expression , which we can perform with omicverse. For different expression analysis, ov change the `gene_id` to `gene_name` of matrix first.
|
| 7 |
+
#
|
| 8 |
+
# Now we can use `PyDEseq2` to perform DESeq2 analysis like R
|
| 9 |
+
#
|
| 10 |
+
# Paper: [PyDESeq2: a python package for bulk RNA-seq differential expression analysis](https://www.biorxiv.org/content/10.1101/2022.12.14.520412v1)
|
| 11 |
+
#
|
| 12 |
+
# Code: https://github.com/owkin/PyDESeq2
|
| 13 |
+
#
|
| 14 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1fZS-v0zdIYkXrEoIAM1X5kPoZVfVvY5h?usp=sharing
|
| 15 |
+
|
| 16 |
+
# In[1]:
|
| 17 |
+
|
| 18 |
+
|
| 19 |
+
import omicverse as ov
|
| 20 |
+
ov.utils.ov_plot_set()
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
# Note that this dataset has not been processed in any way and is only exported by `featureCounts`, and Sequence alignment was performed from the genome file of CRCm39
|
| 24 |
+
|
| 25 |
+
# In[2]:
|
| 26 |
+
|
| 27 |
+
|
| 28 |
+
data=ov.utils.read('https://raw.githubusercontent.com/Starlitnightly/Pyomic/master/sample/counts.txt',index_col=0,header=1)
|
| 29 |
+
#replace the columns `.bam` to ``
|
| 30 |
+
data.columns=[i.split('/')[-1].replace('.bam','') for i in data.columns]
|
| 31 |
+
data.head()
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# ## ID mapping
|
| 35 |
+
#
|
| 36 |
+
# We performed the gene_id mapping by the mapping pair file `GRCm39` downloaded before.
|
| 37 |
+
|
| 38 |
+
# In[ ]:
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
ov.utils.download_geneid_annotation_pair()
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# In[3]:
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
data=ov.bulk.Matrix_ID_mapping(data,'genesets/pair_GRCm39.tsv')
|
| 48 |
+
data.head()
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
# ## Different expression analysis with ov
|
| 52 |
+
#
|
| 53 |
+
# We can do differential expression analysis very simply by ov, simply by providing an expression matrix. To run DEG, we simply need to:
|
| 54 |
+
#
|
| 55 |
+
# - Read the raw count by featureCount or any other qualify methods.
|
| 56 |
+
# - Create an ov DEseq object.
|
| 57 |
+
|
| 58 |
+
# In[4]:
|
| 59 |
+
|
| 60 |
+
|
| 61 |
+
dds=ov.bulk.pyDEG(data)
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# We notes that the gene_name mapping before exist some duplicates, we will process the duplicate indexes to retain only the highest expressed genes
|
| 65 |
+
|
| 66 |
+
# In[5]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
dds.drop_duplicates_index()
|
| 70 |
+
print('... drop_duplicates_index success')
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# Now we can calculate the different expression gene from matrix, we need to input the treatment and control groups
|
| 74 |
+
|
| 75 |
+
# In[6]:
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
treatment_groups=['4-3','4-4']
|
| 79 |
+
control_groups=['1--1','1--2']
|
| 80 |
+
result=dds.deg_analysis(treatment_groups,control_groups,method='DEseq2')
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
# One important thing is that we do not filter out low expression genes when processing DEGs, and in future versions I will consider building in the corresponding processing.
|
| 84 |
+
|
| 85 |
+
# In[7]:
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
print(result.shape)
|
| 89 |
+
result=result.loc[result['log2(BaseMean)']>1]
|
| 90 |
+
print(result.shape)
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
# We also need to set the threshold of Foldchange, we prepare a method named `foldchange_set` to finish. This function automatically calculates the appropriate threshold based on the log2FC distribution, but you can also enter it manually.
|
| 94 |
+
|
| 95 |
+
# In[8]:
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# -1 means automatically calculates
|
| 99 |
+
dds.foldchange_set(fc_threshold=-1,
|
| 100 |
+
pval_threshold=0.05,
|
| 101 |
+
logp_max=10)
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
# ## Visualize the DEG result and specific genes
|
| 105 |
+
#
|
| 106 |
+
# To visualize the DEG result, we use `plot_volcano` to do it. This fuction can visualize the gene interested or high different expression genes. There are some parameters you need to input:
|
| 107 |
+
#
|
| 108 |
+
# - title: The title of volcano
|
| 109 |
+
# - figsize: The size of figure
|
| 110 |
+
# - plot_genes: The genes you interested
|
| 111 |
+
# - plot_genes_num: If you don't have interested genes, you can auto plot it.
|
| 112 |
+
|
| 113 |
+
# In[9]:
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
|
| 117 |
+
plot_genes_num=8,plot_genes_fontsize=12,)
|
| 118 |
+
|
| 119 |
+
|
| 120 |
+
# To visualize the specific genes, we only need to use the `dds.plot_boxplot` function to finish it.
|
| 121 |
+
|
| 122 |
+
# In[10]:
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
dds.plot_boxplot(genes=['Ckap2','Lef1'],treatment_groups=treatment_groups,
|
| 126 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 127 |
+
legend_bbox=(2,0.55))
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# In[11]:
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
dds.plot_boxplot(genes=['Ckap2'],treatment_groups=treatment_groups,
|
| 134 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 135 |
+
legend_bbox=(2,0.55))
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
# ## Pathway enrichment analysis by Pyomic
|
| 139 |
+
#
|
| 140 |
+
# Here we use the `gseapy` package, which included the GSEA analysis and Enrichment. We have optimised the output of the package and given some better looking graph drawing functions
|
| 141 |
+
#
|
| 142 |
+
# Similarly, we need to download the pathway/genesets first. Five genesets we prepare previously, you can use `Pyomic.utils.download_pathway_database()` to download automatically. Besides, you can download the pathway you interested from enrichr: https://maayanlab.cloud/Enrichr/#libraries
|
| 143 |
+
|
| 144 |
+
# In[13]:
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
ov.utils.download_pathway_database()
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
# In[14]:
|
| 151 |
+
|
| 152 |
+
|
| 153 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/WikiPathways_2019_Mouse.txt',organism='Mouse')
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# To perform the GSEA analysis, we need to ranking the genes at first. Using `dds.ranking2gsea` can obtain a ranking gene's matrix sorted by -log10(padj).
|
| 157 |
+
#
|
| 158 |
+
# $Metric=\frac{-log_{10}(padj)}{sign(log2FC)}$
|
| 159 |
+
|
| 160 |
+
# In[15]:
|
| 161 |
+
|
| 162 |
+
|
| 163 |
+
rnk=dds.ranking2gsea()
|
| 164 |
+
|
| 165 |
+
|
| 166 |
+
# We used `ov.bulk.pyGSEA` to construst a GSEA object to perform enrichment.
|
| 167 |
+
|
| 168 |
+
# In[16]:
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
gsea_obj=ov.bulk.pyGSEA(rnk,pathway_dict)
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# In[17]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
enrich_res=gsea_obj.enrichment()
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
# The results are stored in the `enrich_res` attribute.
|
| 181 |
+
|
| 182 |
+
# In[18]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
gsea_obj.enrich_res.head()
|
| 186 |
+
|
| 187 |
+
|
| 188 |
+
# To visualize the enrichment, we use `plot_enrichment` to do.
|
| 189 |
+
# - num: The number of enriched terms to plot. Default is 10.
|
| 190 |
+
# - node_size: A list of integers defining the size of nodes in the plot. Default is [5,10,15].
|
| 191 |
+
# - cax_loc: The location of the colorbar on the plot. Default is 2.
|
| 192 |
+
# - cax_fontsize: The fontsize of the colorbar label. Default is 12.
|
| 193 |
+
# - fig_title: The title of the plot. Default is an empty string.
|
| 194 |
+
# - fig_xlabel: The label of the x-axis. Default is 'Fractions of genes'.
|
| 195 |
+
# - figsize: The size of the plot. Default is (2,4).
|
| 196 |
+
# - cmap: The colormap to use for the plot. Default is 'YlGnBu'.
|
| 197 |
+
|
| 198 |
+
# In[19]:
|
| 199 |
+
|
| 200 |
+
|
| 201 |
+
gsea_obj.plot_enrichment(num=10,node_size=[10,20,30],
|
| 202 |
+
cax_fontsize=12,
|
| 203 |
+
fig_title='Wiki Pathway Enrichment',fig_xlabel='Fractions of genes',
|
| 204 |
+
figsize=(2,4),cmap='YlGnBu',
|
| 205 |
+
text_knock=2,text_maxsize=30,
|
| 206 |
+
cax_loc=[2.5, 0.45, 0.5, 0.02],
|
| 207 |
+
bbox_to_anchor_used=(-0.25, -13),node_diameter=10,)
|
| 208 |
+
|
| 209 |
+
|
| 210 |
+
# Not only the basic analysis, pyGSEA also help us to visualize the term with Ranked and Enrichment Score.
|
| 211 |
+
#
|
| 212 |
+
# We can select the number of term to plot, which stored in `gsea_obj.enrich_res.index`, the `0` is `Complement and Coagulation Cascades WP449` and the `1` is `Matrix Metalloproteinases WP441`
|
| 213 |
+
|
| 214 |
+
# In[20]:
|
| 215 |
+
|
| 216 |
+
|
| 217 |
+
gsea_obj.enrich_res.index[:5]
|
| 218 |
+
|
| 219 |
+
|
| 220 |
+
# We can set the `gene_set_title` to change the title of GSEA plot
|
| 221 |
+
|
| 222 |
+
# In[22]:
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
fig=gsea_obj.plot_gsea(term_num=1,
|
| 226 |
+
gene_set_title='Matrix Metalloproteinases',
|
| 227 |
+
figsize=(3,4),
|
| 228 |
+
cmap='RdBu_r',
|
| 229 |
+
title_fontsize=14,
|
| 230 |
+
title_y=0.95)
|
| 231 |
+
|
| 232 |
+
|
| 233 |
+
# In[ ]:
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
|
| 237 |
+
|
ovrawm/t_gptanno.txt
ADDED
|
@@ -0,0 +1,330 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Automatic cell type annotation with GPT/Other
|
| 5 |
+
#
|
| 6 |
+
# GPTCelltype, an open-source R software package to facilitate cell type annotation by GPT-4.
|
| 7 |
+
#
|
| 8 |
+
# We made three improvements in integrating the `GPTCelltype` algorithm in OmicVerse:
|
| 9 |
+
#
|
| 10 |
+
# - **Native support for Python**: Since GPTCelltype is an R language package, in order to make it conform to scverse's anndata ecosystem, we have rewritten the whole function so that it works perfectly under Python.
|
| 11 |
+
# - **More model support**: We provide more big models to choose from outside of Openai, e.g. Qwen(通义千问), Kimi, and more model support is available through the parameter `base_url` or `model_name`.
|
| 12 |
+
#
|
| 13 |
+
# If you found this tutorial helpful, please cite GPTCelltype and OmicVerse:
|
| 14 |
+
#
|
| 15 |
+
# Hou, W. and Ji, Z., 2023. Reference-free and cost-effective automated cell type annotation with GPT-4 in single-cell RNA-seq analysis. [Nature Methods, 2024 March 25](https://link.springer.com/article/10.1038/s41592-024-02235-4?utm_source=rct_congratemailt&utm_medium=email&utm_campaign=oa_20240325&utm_content=10.1038/s41592-024-02235-4).
|
| 16 |
+
|
| 17 |
+
# In[3]:
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
import omicverse as ov
|
| 21 |
+
print(f'omicverse version:{ov.__version__}')
|
| 22 |
+
import scanpy as sc
|
| 23 |
+
print(f'scanpy version:{sc.__version__}')
|
| 24 |
+
ov.ov_plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# ## Loading data
|
| 28 |
+
#
|
| 29 |
+
# The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics ([here](http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz) from this [webpage](https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k)). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
|
| 30 |
+
#
|
| 31 |
+
|
| 32 |
+
# In[21]:
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# !mkdir data
|
| 36 |
+
# !wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 37 |
+
# !cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 38 |
+
# !mkdir write
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
# Read in the count matrix into an AnnData object, which holds many slots for annotations and different representations of the data. It also comes with its own HDF5-based file format: `.h5ad`.
|
| 42 |
+
|
| 43 |
+
# In[3]:
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
adata = sc.read_10x_mtx(
|
| 47 |
+
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
|
| 48 |
+
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
|
| 49 |
+
cache=True) # write a cache file for faster subsequent reading
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
# ## Data preprocessing
|
| 53 |
+
#
|
| 54 |
+
# Here, we use `ov.single.scanpy_lazy` to preprocess the raw data of scRNA-seq, it included filter the doublets cells, normalizing counts per cell, log1p, extracting highly variable genes, and cluster of cells calculation.
|
| 55 |
+
#
|
| 56 |
+
# But if you want to experience step-by-step preprocessing, we also provide more detailed preprocessing steps here, please refer to our [preprocess chapter](https://omicverse.readthedocs.io/en/latest/Tutorials-single/t_preprocess/) for a detailed explanation.
|
| 57 |
+
#
|
| 58 |
+
# We stored the raw counts in `count` layers, and the raw data in `adata.raw.to_adata()`.
|
| 59 |
+
|
| 60 |
+
# In[ ]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
#adata=ov.single.scanpy_lazy(adata)
|
| 64 |
+
|
| 65 |
+
#quantity control
|
| 66 |
+
adata=ov.pp.qc(adata,
|
| 67 |
+
tresh={'mito_perc': 0.05, 'nUMIs': 500, 'detected_genes': 250})
|
| 68 |
+
#normalize and high variable genes (HVGs) calculated
|
| 69 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 70 |
+
|
| 71 |
+
#save the whole genes and filter the non-HVGs
|
| 72 |
+
adata.raw = adata
|
| 73 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 74 |
+
|
| 75 |
+
#scale the adata.X
|
| 76 |
+
ov.pp.scale(adata)
|
| 77 |
+
|
| 78 |
+
#Dimensionality Reduction
|
| 79 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 80 |
+
|
| 81 |
+
#Neighbourhood graph construction
|
| 82 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 83 |
+
use_rep='scaled|original|X_pca')
|
| 84 |
+
|
| 85 |
+
#clusters
|
| 86 |
+
sc.tl.leiden(adata)
|
| 87 |
+
|
| 88 |
+
#find marker
|
| 89 |
+
sc.tl.dendrogram(adata,'leiden',use_rep='scaled|original|X_pca')
|
| 90 |
+
sc.tl.rank_genes_groups(adata, 'leiden', use_rep='scaled|original|X_pca',
|
| 91 |
+
method='wilcoxon',use_raw=False,)
|
| 92 |
+
|
| 93 |
+
#Dimensionality Reduction for visualization(X_mde=X_umap+GPU)
|
| 94 |
+
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 95 |
+
adata
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# In[5]:
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
ov.pl.embedding(adata,
|
| 102 |
+
basis='X_mde',
|
| 103 |
+
color=['leiden'],
|
| 104 |
+
legend_loc='on data',
|
| 105 |
+
frameon='small',
|
| 106 |
+
legend_fontoutline=2,
|
| 107 |
+
palette=ov.utils.palette()[14:],
|
| 108 |
+
)
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
# ## GPT Celltype
|
| 112 |
+
#
|
| 113 |
+
# gptcelltype supports dictionary format input, we provide `omicverse.single.get_celltype_marker` to get the marker genes for each cell type as a dictionary.
|
| 114 |
+
|
| 115 |
+
# #### Using genes manually
|
| 116 |
+
#
|
| 117 |
+
# We can manually define a dictionary to determine the accuracy of the output
|
| 118 |
+
|
| 119 |
+
# In[25]:
|
| 120 |
+
|
| 121 |
+
|
| 122 |
+
import os
|
| 123 |
+
all_markers={'cluster1':['CD3D','CD3E'],
|
| 124 |
+
'cluster2':['MS4A1']}
|
| 125 |
+
|
| 126 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 127 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 128 |
+
model='qwen-plus', provider='qwen',
|
| 129 |
+
topgenenumber=5)
|
| 130 |
+
result
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
# #### Get Genes for Each Cluster Automatically
|
| 134 |
+
#
|
| 135 |
+
|
| 136 |
+
# In[14]:
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
all_markers=ov.single.get_celltype_marker(adata,clustertype='leiden',rank=True,
|
| 140 |
+
key='rank_genes_groups',
|
| 141 |
+
foldchange=2,topgenenumber=5)
|
| 142 |
+
all_markers
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
# ### Option 1. Through OpenAI API (`provider` or `base_url`)
|
| 146 |
+
#
|
| 147 |
+
# Use `ov.single.gptcelltype` function to annotate cell types.
|
| 148 |
+
#
|
| 149 |
+
# You can simply set `provider` (or `base_url`) and `provider` parameters to provide the function with base url and the exact model which are required for model calling.
|
| 150 |
+
|
| 151 |
+
# In[17]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
import os
|
| 155 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 156 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 157 |
+
model='qwen-plus', provider='qwen',
|
| 158 |
+
topgenenumber=5)
|
| 159 |
+
result
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
# We can keep only the cell type of the output and remove other irrelevant information.
|
| 163 |
+
|
| 164 |
+
# In[18]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
new_result={}
|
| 168 |
+
for key in result.keys():
|
| 169 |
+
new_result[key]=result[key].split(': ')[-1].split(' (')[0].split('. ')[1]
|
| 170 |
+
new_result
|
| 171 |
+
|
| 172 |
+
|
| 173 |
+
# In[19]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
adata.obs['gpt_celltype'] = adata.obs['leiden'].map(new_result).astype('category')
|
| 177 |
+
|
| 178 |
+
|
| 179 |
+
# In[20]:
|
| 180 |
+
|
| 181 |
+
|
| 182 |
+
ov.pl.embedding(adata,
|
| 183 |
+
basis='X_mde',
|
| 184 |
+
color=['leiden','gpt_celltype'],
|
| 185 |
+
legend_loc='on data',
|
| 186 |
+
frameon='small',
|
| 187 |
+
legend_fontoutline=2,
|
| 188 |
+
palette=ov.utils.palette()[14:],
|
| 189 |
+
)
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
# ## More models
|
| 193 |
+
#
|
| 194 |
+
# Our implementation of `gptcelltype` in `omicverse` supports almost all large language models that support the `openai` api format.
|
| 195 |
+
|
| 196 |
+
# In[27]:
|
| 197 |
+
|
| 198 |
+
|
| 199 |
+
all_markers={'cluster1':['CD3D','CD3E'],
|
| 200 |
+
'cluster2':['MS4A1']}
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
# ### Openai
|
| 204 |
+
#
|
| 205 |
+
# The OpenAI API uses API keys for authentication. You can create API keys at a user or service account level. Service accounts are tied to a "bot" individual and should be used to provision access for production systems. Each API key can be scoped to one of the following,
|
| 206 |
+
#
|
| 207 |
+
# - [User keys](https://platform.openai.com/account/api-keys) - Our legacy keys. Provides access to all organizations and all projects that user has been added to; access API Keys to view your available keys. We highly advise transitioning to project keys for best security practices, although access via this method is currently still supported.
|
| 208 |
+
#
|
| 209 |
+
# - Please select the model you need to use: [list of supported models](https://platform.openai.com/docs/models).
|
| 210 |
+
#
|
| 211 |
+
|
| 212 |
+
# In[28]:
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 216 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 217 |
+
model='gpt-4o', provider='openai',
|
| 218 |
+
topgenenumber=5)
|
| 219 |
+
result
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
# ### Qwen(通义千问)
|
| 223 |
+
#
|
| 224 |
+
# - Enabled DashScope service and obtained API-KEY: [Enabled DashScope and created API-KEY](https://help.aliyun.com/zh/dashscope/developer-reference/activate-dashscope-and-create-an-api-key).
|
| 225 |
+
#
|
| 226 |
+
# - We recommend you to configure API-KEY in environment variable to reduce the risk of API-KEY leakage, please refer to Configuring API-KEY through Environment Variable for the configuration method, you can also configure API-KEY in code, but the risk of leakage will be increased.
|
| 227 |
+
#
|
| 228 |
+
# - Please select the model you need to use: [list of supported models](https://help.aliyun.com/zh/dashscope/developer-reference/compatibility-of-openai-with-dashscope/?spm=a2c4g.11186623.0.i6#eadfc13038jd5).
|
| 229 |
+
#
|
| 230 |
+
# **简体中文**
|
| 231 |
+
#
|
| 232 |
+
# - 已开通灵积模型服务并获得API-KEY:[开通DashScope并创建API-KEY](https://help.aliyun.com/zh/dashscope/developer-reference/activate-dashscope-and-create-an-api-key)。
|
| 233 |
+
#
|
| 234 |
+
# - 我们推荐您将API-KEY配置到环境变量中以降低API-KEY的泄漏风险,配置方法可参考通过环境变量配置API-KEY。您也可以在代码中配置API-KEY,但是泄漏风险会提高。
|
| 235 |
+
#
|
| 236 |
+
# - 请选择您需要使用的模型:[支持的模型列表](https://help.aliyun.com/zh/dashscope/developer-reference/compatibility-of-openai-with-dashscope/?spm=a2c4g.11186623.0.i6#eadfc13038jd5)。
|
| 237 |
+
#
|
| 238 |
+
|
| 239 |
+
# In[26]:
|
| 240 |
+
|
| 241 |
+
|
| 242 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 243 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 244 |
+
model='qwen-plus', provider='qwen',
|
| 245 |
+
topgenenumber=5)
|
| 246 |
+
result
|
| 247 |
+
|
| 248 |
+
|
| 249 |
+
# ### Kimi(月之暗面)
|
| 250 |
+
#
|
| 251 |
+
# - You will need a Dark Side of the Moon API key to use our service. You can create an API key in [Console](https://platform.moonshot.cn/console).
|
| 252 |
+
#
|
| 253 |
+
# - Please select the model you need to use: [List of supported models](https://platform.moonshot.cn/docs/pricing#%E4%BA%A7%E5%93%81%E5%AE%9A%E4%BB%B7)
|
| 254 |
+
#
|
| 255 |
+
# **简体中文**
|
| 256 |
+
#
|
| 257 |
+
# - 你需要一个 月之暗面的 API 密钥��使用我们的服务。你可以在[控制台](https://platform.moonshot.cn/console)中创建一个 API 密钥。
|
| 258 |
+
#
|
| 259 |
+
# - 请选择您需要使用的模型:[支持的模型列表](https://platform.moonshot.cn/docs/pricing#%E4%BA%A7%E5%93%81%E5%AE%9A%E4%BB%B7)
|
| 260 |
+
|
| 261 |
+
# In[28]:
|
| 262 |
+
|
| 263 |
+
|
| 264 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 265 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 266 |
+
model='moonshot-v1-8k', provider='kimi',
|
| 267 |
+
topgenenumber=5)
|
| 268 |
+
result
|
| 269 |
+
|
| 270 |
+
|
| 271 |
+
# #### Other Models
|
| 272 |
+
#
|
| 273 |
+
# You can manually set the `base_url` parameter to specify other models that need to be used, note that the model needs to support Openai's parameters. Three examples are provided here (when you specify the `base_url` parameter, the `provider` parameter will be invalid):
|
| 274 |
+
#
|
| 275 |
+
# ```python
|
| 276 |
+
# if provider == 'openai':
|
| 277 |
+
# base_url = "https://api.openai.com/v1/"
|
| 278 |
+
# elif provider == 'kimi':
|
| 279 |
+
# base_url = "https://api.moonshot.cn/v1"
|
| 280 |
+
# elif provider == 'qwen':
|
| 281 |
+
# base_url = "https://dashscope.aliyuncs.com/compatible-mode/v1"
|
| 282 |
+
# ```
|
| 283 |
+
|
| 284 |
+
# In[30]:
|
| 285 |
+
|
| 286 |
+
|
| 287 |
+
os.environ['AGI_API_KEY'] = 'sk-**' # Replace with your actual API key
|
| 288 |
+
result = ov.single.gptcelltype(all_markers, tissuename='PBMC', speciename='human',
|
| 289 |
+
model='moonshot-v1-8k', base_url="https://api.moonshot.cn/v1",
|
| 290 |
+
topgenenumber=5)
|
| 291 |
+
result
|
| 292 |
+
|
| 293 |
+
|
| 294 |
+
# ### Option 2. Through Local LLM (`model_name`)
|
| 295 |
+
|
| 296 |
+
# Use `ov.single.gptcelltype_local` function to annotate cell types.
|
| 297 |
+
#
|
| 298 |
+
# You can simply set the `model_name` parameter.
|
| 299 |
+
|
| 300 |
+
# In[5]:
|
| 301 |
+
|
| 302 |
+
|
| 303 |
+
anno_model = 'path/to/your/local/LLM' # '~/models/Qwen2-7B-Instruct'
|
| 304 |
+
|
| 305 |
+
result = ov.single.gptcelltype_local(all_markers, tissuename='PBMC', speciename='human',
|
| 306 |
+
model_name=anno_model, topgenenumber=5)
|
| 307 |
+
result
|
| 308 |
+
|
| 309 |
+
|
| 310 |
+
# Note that you may encounter network problems that prevent you from downloading LLMs.
|
| 311 |
+
#
|
| 312 |
+
# In this case, please refer to https://zhuanlan.zhihu.com/p/663712983
|
| 313 |
+
|
| 314 |
+
# In[ ]:
|
| 315 |
+
|
| 316 |
+
|
| 317 |
+
|
| 318 |
+
|
| 319 |
+
|
| 320 |
+
# In[ ]:
|
| 321 |
+
|
| 322 |
+
|
| 323 |
+
|
| 324 |
+
|
| 325 |
+
|
| 326 |
+
# In[ ]:
|
| 327 |
+
|
| 328 |
+
|
| 329 |
+
|
| 330 |
+
|
ovrawm/t_mapping.txt
ADDED
|
@@ -0,0 +1,191 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Mapping single-cell profile onto spatial profile
|
| 5 |
+
#
|
| 6 |
+
# Tangram is a method for mapping single-cell (or single-nucleus) gene expression data onto spatial gene expression data. Tangram takes as input a single-cell dataset and a spatial dataset, collected from the same anatomical region/tissue type. Via integration, Tangram creates new spatial data by aligning the scRNAseq profiles in space. This allows to project every annotation in the scRNAseq (e.g. cell types, program usage) on space.
|
| 7 |
+
#
|
| 8 |
+
# The most common application of Tangram is to resolve cell types in space. Another usage is to correct gene expression from spatial data: as scRNA-seq data are less prone to dropout than (e.g.) Visium or Slide-seq, the “new” spatial data generated by Tangram resolve many more genes. As a result, we can visualize program usage in space, which can be used for ligand-receptor pair discovery or, more generally, cell-cell communication mechanisms. If cell segmentation is available, Tangram can be also used for deconvolution of spatial data. If your single cell are multimodal, Tangram can be used to spatially resolve other modalities, such as chromatin accessibility.
|
| 9 |
+
#
|
| 10 |
+
# Biancalani, T., Scalia, G., Buffoni, L. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat Methods 18, 1352–1362 (2021). https://doi.org/10.1038/s41592-021-01264-7
|
| 11 |
+
#
|
| 12 |
+
# 
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 19 |
+
import scanpy as sc
|
| 20 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 21 |
+
ov.utils.ov_plot_set()
|
| 22 |
+
|
| 23 |
+
|
| 24 |
+
# ## Prepared scRNA-seq
|
| 25 |
+
#
|
| 26 |
+
# Published scRNA-seq datasets of lymph nodes have typically lacked an adequate representation of germinal centre-associated immune cell populations due to age of patient donors. We, therefore, include scRNA-seq datasets spanning lymph nodes, spleen and tonsils in our single-cell reference to ensure that we captured the full diversity of immune cell states likely to exist in the spatial transcriptomic dataset.
|
| 27 |
+
#
|
| 28 |
+
# Here we download this dataset, import into anndata and change variable names to ENSEMBL gene identifiers.
|
| 29 |
+
#
|
| 30 |
+
# Link: https://cell2location.cog.sanger.ac.uk/paper/integrated_lymphoid_organ_scrna/RegressionNBV4Torch_57covariates_73260cells_10237genes/sc.h5ad
|
| 31 |
+
|
| 32 |
+
# In[2]:
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
adata_sc=ov.read('data/sc.h5ad')
|
| 36 |
+
import matplotlib.pyplot as plt
|
| 37 |
+
fig, ax = plt.subplots(figsize=(3,3))
|
| 38 |
+
ov.utils.embedding(
|
| 39 |
+
adata_sc,
|
| 40 |
+
basis="X_umap",
|
| 41 |
+
color=['Subset'],
|
| 42 |
+
title='Subset',
|
| 43 |
+
frameon='small',
|
| 44 |
+
#ncols=1,
|
| 45 |
+
wspace=0.65,
|
| 46 |
+
#palette=ov.utils.pyomic_palette()[11:],
|
| 47 |
+
show=False,
|
| 48 |
+
ax=ax
|
| 49 |
+
)
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
# For data quality control and preprocessing, we can easily use omicverse's own preprocessing functions to do so
|
| 53 |
+
|
| 54 |
+
# In[3]:
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
print("RAW",adata_sc.X.max())
|
| 58 |
+
adata_sc=ov.pp.preprocess(adata_sc,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 59 |
+
adata_sc.raw = adata_sc
|
| 60 |
+
adata_sc = adata_sc[:, adata_sc.var.highly_variable_features]
|
| 61 |
+
print("Normalize",adata_sc.X.max())
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# ## Prepared stRNA-seq
|
| 65 |
+
#
|
| 66 |
+
# First let’s read spatial Visium data from 10X Space Ranger output. Here we use lymph node data generated by 10X and presented in [Kleshchevnikov et al (section 4, Fig 4)](https://www.biorxiv.org/content/10.1101/2020.11.15.378125v1). This dataset can be conveniently downloaded and imported using scanpy. See [this tutorial](https://cell2location.readthedocs.io/en/latest/notebooks/cell2location_short_demo.html) for a more extensive and practical example of data loading (multiple visium samples).
|
| 67 |
+
|
| 68 |
+
# In[5]:
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
adata = sc.datasets.visium_sge(sample_id="V1_Human_Lymph_Node")
|
| 72 |
+
adata.obs['sample'] = list(adata.uns['spatial'].keys())[0]
|
| 73 |
+
adata.var_names_make_unique()
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
# We used the same pre-processing steps as for scRNA-seq
|
| 77 |
+
#
|
| 78 |
+
# <div class="admonition warning">
|
| 79 |
+
# <p class="admonition-title">Note</p>
|
| 80 |
+
# <p>
|
| 81 |
+
# We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code.
|
| 82 |
+
# </p>
|
| 83 |
+
# </div>
|
| 84 |
+
#
|
| 85 |
+
# ```python
|
| 86 |
+
# #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 87 |
+
# #adata.raw = adata
|
| 88 |
+
# #adata = adata[:, adata.var.highly_variable_features]
|
| 89 |
+
# ```
|
| 90 |
+
|
| 91 |
+
# In[6]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
sc.pp.calculate_qc_metrics(adata, inplace=True)
|
| 95 |
+
adata = adata[:,adata.var['total_counts']>100]
|
| 96 |
+
adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",)
|
| 97 |
+
adata.raw = adata
|
| 98 |
+
adata = adata[:, adata.var.space_variable_features]
|
| 99 |
+
adata_sp=adata.copy()
|
| 100 |
+
adata_sp
|
| 101 |
+
|
| 102 |
+
|
| 103 |
+
# ## Tangram model
|
| 104 |
+
#
|
| 105 |
+
# Tangram is a Python package, written in PyTorch and based on scanpy, for mapping single-cell (or single-nucleus) gene expression data onto spatial gene expression data. The single-cell dataset and the spatial dataset should be collected from the same anatomical region/tissue type, ideally from a biological replicate, and need to share a set of genes.
|
| 106 |
+
#
|
| 107 |
+
# We can use `omicverse.space.Tangram` to apply the Tangram model.
|
| 108 |
+
|
| 109 |
+
# In[7]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
tg=ov.space.Tangram(adata_sc,adata_sp,clusters='Subset')
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# The function maps iteratively as specified by num_epochs. We typically interrupt mapping after the score plateaus.
|
| 116 |
+
# - The score measures the similarity between the gene expression of the mapped cells vs spatial data on the training genes.
|
| 117 |
+
# - The default mapping mode is mode=`cells`, which is recommended to run on a GPU.
|
| 118 |
+
# - Alternatively, one can specify mode=`clusters` which averages the single cells beloning to the same cluster (pass annotations via cluster_label). This is faster, and is our chioce when scRNAseq and spatial data come from different specimens.
|
| 119 |
+
# - If you wish to run Tangram with a GPU, set device=`cuda:0` otherwise use the set device=`cpu`.
|
| 120 |
+
# - density_prior specifies the cell density within each spatial voxel. Use uniform if the spatial voxels are at single cell resolution (ie MERFISH). The default value, rna_count_based, assumes that cell density is proportional to the number of RNA molecules
|
| 121 |
+
|
| 122 |
+
# In[8]:
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
tg.train(mode="clusters",num_epochs=500,device="cuda:0")
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
# We can use `tg.cell2location()` to get the cell location in spatial spots.
|
| 129 |
+
|
| 130 |
+
# In[9]:
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
adata_plot=tg.cell2location()
|
| 134 |
+
adata_plot.obs.columns
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
# In[10]:
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
annotation_list=['B_Cycling', 'B_GC_LZ', 'T_CD4+_TfH_GC', 'FDC',
|
| 141 |
+
'B_naive', 'T_CD4+_naive', 'B_plasma', 'Endo']
|
| 142 |
+
|
| 143 |
+
sc.pl.spatial(adata_plot, cmap='magma',
|
| 144 |
+
# show first 8 cell types
|
| 145 |
+
color=annotation_list,
|
| 146 |
+
ncols=4, size=1.3,
|
| 147 |
+
img_key='hires',
|
| 148 |
+
# limit color scale at 99.2% quantile of cell abundance
|
| 149 |
+
#vmin=0, vmax='p99.2'
|
| 150 |
+
)
|
| 151 |
+
|
| 152 |
+
|
| 153 |
+
# In[11]:
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
color_dict=dict(zip(adata_sc.obs['Subset'].cat.categories,
|
| 157 |
+
adata_sc.uns['Subset_colors']))
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
# In[21]:
|
| 161 |
+
|
| 162 |
+
|
| 163 |
+
import matplotlib as mpl
|
| 164 |
+
clust_labels = annotation_list[:5]
|
| 165 |
+
clust_col = ['' + str(i) for i in clust_labels] # in case column names differ from labels
|
| 166 |
+
|
| 167 |
+
with mpl.rc_context({'figure.figsize': (8, 8),'axes.grid': False}):
|
| 168 |
+
fig = ov.pl.plot_spatial(
|
| 169 |
+
adata=adata_plot,
|
| 170 |
+
# labels to show on a plot
|
| 171 |
+
color=clust_col, labels=clust_labels,
|
| 172 |
+
show_img=True,
|
| 173 |
+
# 'fast' (white background) or 'dark_background'
|
| 174 |
+
style='fast',
|
| 175 |
+
# limit color scale at 99.2% quantile of cell abundance
|
| 176 |
+
max_color_quantile=0.992,
|
| 177 |
+
# size of locations (adjust depending on figure size)
|
| 178 |
+
circle_diameter=3,
|
| 179 |
+
reorder_cmap = [#0,
|
| 180 |
+
1,2,3,4,6], #['yellow', 'orange', 'blue', 'green', 'purple', 'grey', 'white'],
|
| 181 |
+
colorbar_position='right',
|
| 182 |
+
palette=color_dict
|
| 183 |
+
)
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# In[ ]:
|
| 188 |
+
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
|
ovrawm/t_metacells.txt
ADDED
|
@@ -0,0 +1,249 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Inference of MetaCell from Single-Cell RNA-seq
|
| 5 |
+
#
|
| 6 |
+
# Metacells are cell groupings derived from single-cell sequencing data that represent highly granular, distinct cell states. Here, we present single-cell aggregation of cell-states (SEACells), an algorithm for identifying metacells; overcoming the sparsity of single-cell data, while retaining heterogeneity obscured by traditional cell clustering.
|
| 7 |
+
#
|
| 8 |
+
# Paper: [SEACells: Inference of transcriptional and epigenomic cellular states from single-cell genomics data](https://www.nature.com/articles/s41587-023-01716-9)
|
| 9 |
+
#
|
| 10 |
+
# Code: https://github.com/dpeerlab/SEACells
|
| 11 |
+
#
|
| 12 |
+
|
| 13 |
+
# In[1]:
|
| 14 |
+
|
| 15 |
+
|
| 16 |
+
import omicverse as ov
|
| 17 |
+
import scanpy as sc
|
| 18 |
+
import scvelo as scv
|
| 19 |
+
|
| 20 |
+
ov.plot_set()
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
# ## Data preprocessed
|
| 24 |
+
#
|
| 25 |
+
# We need to normalized and scale the data at first.
|
| 26 |
+
|
| 27 |
+
# In[3]:
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
adata = scv.datasets.pancreas()
|
| 31 |
+
adata
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# In[4]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
#quantity control
|
| 38 |
+
adata=ov.pp.qc(adata,
|
| 39 |
+
tresh={'mito_perc': 0.20, 'nUMIs': 500, 'detected_genes': 250},
|
| 40 |
+
mt_startswith='mt-')
|
| 41 |
+
#normalize and high variable genes (HVGs) calculated
|
| 42 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 43 |
+
|
| 44 |
+
#save the whole genes and filter the non-HVGs
|
| 45 |
+
adata.raw = adata
|
| 46 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 47 |
+
|
| 48 |
+
#scale the adata.X
|
| 49 |
+
ov.pp.scale(adata)
|
| 50 |
+
|
| 51 |
+
#Dimensionality Reduction
|
| 52 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
# ## Constructing a metacellular object
|
| 56 |
+
#
|
| 57 |
+
# We can use `ov.single.MetaCell` to construct a metacellular object to train the SEACells model, the arguments can be found in below.
|
| 58 |
+
#
|
| 59 |
+
# - :param ad: (AnnData) annotated data matrix
|
| 60 |
+
# - :param build_kernel_on: (str) key corresponding to matrix in ad.obsm which is used to compute kernel for metacells
|
| 61 |
+
# Typically 'X_pca' for scRNA or 'X_svd' for scATAC
|
| 62 |
+
# - :param n_SEACells: (int) number of SEACells to compute
|
| 63 |
+
# - :param use_gpu: (bool) whether to use GPU for computation
|
| 64 |
+
# - :param verbose: (bool) whether to suppress verbose program logging
|
| 65 |
+
# - :param n_waypoint_eigs: (int) number of eigenvectors to use for waypoint initialization
|
| 66 |
+
# - :param n_neighbors: (int) number of nearest neighbors to use for graph construction
|
| 67 |
+
# - :param convergence_epsilon: (float) convergence threshold for Franke-Wolfe algorithm
|
| 68 |
+
# - :param l2_penalty: (float) L2 penalty for Franke-Wolfe algorithm
|
| 69 |
+
# - :param max_franke_wolfe_iters: (int) maximum number of iterations for Franke-Wolfe algorithm
|
| 70 |
+
# - :param use_sparse: (bool) whether to use sparse matrix operations. Currently only supported for CPU implementation.
|
| 71 |
+
|
| 72 |
+
# In[5]:
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
meta_obj=ov.single.MetaCell(adata,use_rep='scaled|original|X_pca',
|
| 76 |
+
n_metacells=None,
|
| 77 |
+
use_gpu='cuda:0')
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
# In[6]:
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
get_ipython().run_cell_magic('time', '', 'meta_obj.initialize_archetypes()\n')
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# ## Train and save the model
|
| 87 |
+
|
| 88 |
+
# In[7]:
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
get_ipython().run_cell_magic('time', '', 'meta_obj.train(min_iter=10, max_iter=50)\n')
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
# In[9]:
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
meta_obj.save('seacells/model.pkl')
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# In[6]:
|
| 101 |
+
|
| 102 |
+
|
| 103 |
+
meta_obj.load('seacells/model.pkl')
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
# ## Predicted the metacells
|
| 107 |
+
#
|
| 108 |
+
# we can use `predicted` to predicted the metacells of raw scRNA-seq data. There are two method can be selected, one is `soft`, the other is `hard`.
|
| 109 |
+
#
|
| 110 |
+
# In the `soft` method, Aggregates cells within each SEACell, summing over all raw data x assignment weight for all cells belonging to a SEACell. Data is un-normalized and pseudo-raw aggregated counts are stored in .layers['raw']. Attributes associated with variables (.var) are copied over, but relevant per SEACell attributes must be manually copied, since certain attributes may need to be summed, or averaged etc, depending on the attribute.
|
| 111 |
+
#
|
| 112 |
+
# In the `hard` method, Aggregates cells within each SEACell, summing over all raw data for all cells belonging to a SEACell. Data is unnormalized and raw aggregated counts are stored .layers['raw']. Attributes associated with variables (.var) are copied over, but relevant per SEACell attributes must be manually copied, since certain attributes may need to be summed, or averaged etc, depending on the attribute.
|
| 113 |
+
|
| 114 |
+
# In[10]:
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
ad=meta_obj.predicted(method='soft',celltype_label='clusters',
|
| 118 |
+
summarize_layer='lognorm')
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# ## Benchmarking
|
| 122 |
+
#
|
| 123 |
+
# Benchmarking metrics were computed for each metacell for all combinations of data modality, dataset and method. Cell type purity was used to assess the quality of PBMC metacells. Methods were compared using the Wilcoxon rank-sum test. We note that this test might possibly inflate significance due to the dependency between metacells, but it nonetheless provides an estimate of the direction of difference. Top-performing metacell approaches should have scores that are low on compactness, high on separation and high on cell type purity
|
| 124 |
+
|
| 125 |
+
# In[11]:
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
SEACell_purity = meta_obj.compute_celltype_purity('clusters')
|
| 129 |
+
separation = meta_obj.separation(use_rep='scaled|original|X_pca',nth_nbr=1)
|
| 130 |
+
compactness = meta_obj.compactness(use_rep='scaled|original|X_pca')
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
# In[12]:
|
| 134 |
+
|
| 135 |
+
|
| 136 |
+
import seaborn as sns
|
| 137 |
+
import matplotlib.pyplot as plt
|
| 138 |
+
ov.plot_set()
|
| 139 |
+
fig, axes = plt.subplots(1,3,figsize=(4,4))
|
| 140 |
+
sns.boxplot(data=SEACell_purity, y='clusters_purity',ax=axes[0],
|
| 141 |
+
color=ov.utils.blue_color[3])
|
| 142 |
+
sns.boxplot(data=compactness, y='compactness',ax=axes[1],
|
| 143 |
+
color=ov.utils.blue_color[4])
|
| 144 |
+
sns.boxplot(data=separation, y='separation',ax=axes[2],
|
| 145 |
+
color=ov.utils.blue_color[4])
|
| 146 |
+
plt.tight_layout()
|
| 147 |
+
plt.suptitle('Evaluate of MetaCells',fontsize=13,y=1.05)
|
| 148 |
+
for ax in axes:
|
| 149 |
+
ax.grid(False)
|
| 150 |
+
ax.spines['top'].set_visible(False)
|
| 151 |
+
ax.spines['right'].set_visible(False)
|
| 152 |
+
ax.spines['bottom'].set_visible(True)
|
| 153 |
+
ax.spines['left'].set_visible(True)
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# In[13]:
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
import matplotlib.pyplot as plt
|
| 160 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 161 |
+
ov.pl.embedding(
|
| 162 |
+
meta_obj.adata,
|
| 163 |
+
basis="X_umap",
|
| 164 |
+
color=['clusters'],
|
| 165 |
+
frameon='small',
|
| 166 |
+
title="Meta cells",
|
| 167 |
+
#legend_loc='on data',
|
| 168 |
+
legend_fontsize=14,
|
| 169 |
+
legend_fontoutline=2,
|
| 170 |
+
size=10,
|
| 171 |
+
ax=ax,
|
| 172 |
+
alpha=0.2,
|
| 173 |
+
#legend_loc='',
|
| 174 |
+
add_outline=False,
|
| 175 |
+
#add_outline=True,
|
| 176 |
+
outline_color='black',
|
| 177 |
+
outline_width=1,
|
| 178 |
+
show=False,
|
| 179 |
+
#palette=ov.utils.blue_color[:],
|
| 180 |
+
#legend_fontweight='normal'
|
| 181 |
+
)
|
| 182 |
+
ov.single.plot_metacells(ax,meta_obj.adata,color='#CB3E35',
|
| 183 |
+
)
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
# ## Get the raw obs value from adata
|
| 187 |
+
#
|
| 188 |
+
# There are times when we compute some floating point type data such as pseudotime on the raw single cell data. We want to get the result of the original data on the metacell, in this case, we can use the `ov.single` function to get it.
|
| 189 |
+
#
|
| 190 |
+
# Note that the type parameter supports `str`,`max`,`min`,`mean`.
|
| 191 |
+
|
| 192 |
+
# In[14]:
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
ov.single.get_obs_value(ad,adata,groupby='S_score',
|
| 196 |
+
type='mean')
|
| 197 |
+
ad.obs.head()
|
| 198 |
+
|
| 199 |
+
|
| 200 |
+
# ## Visualize the MetaCells
|
| 201 |
+
|
| 202 |
+
# In[15]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
import scanpy as sc
|
| 206 |
+
ad.raw=ad.copy()
|
| 207 |
+
sc.pp.highly_variable_genes(ad, n_top_genes=2000, inplace=True)
|
| 208 |
+
ad=ad[:,ad.var.highly_variable]
|
| 209 |
+
|
| 210 |
+
|
| 211 |
+
# In[16]:
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
ov.pp.scale(ad)
|
| 215 |
+
ov.pp.pca(ad,layer='scaled',n_pcs=30)
|
| 216 |
+
ov.pp.neighbors(ad, n_neighbors=15, n_pcs=20,
|
| 217 |
+
use_rep='scaled|original|X_pca')
|
| 218 |
+
|
| 219 |
+
|
| 220 |
+
# In[17]:
|
| 221 |
+
|
| 222 |
+
|
| 223 |
+
ov.pp.umap(ad)
|
| 224 |
+
|
| 225 |
+
|
| 226 |
+
# We want the metacells to take on the same colours as the original data, a noteworthy fact is that the colours of the original data are stored in the `adata.uns['_colors']`
|
| 227 |
+
|
| 228 |
+
# In[18]:
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
ad.obs['celltype']=ad.obs['celltype'].astype('category')
|
| 232 |
+
ad.obs['celltype']=ad.obs['celltype'].cat.reorder_categories(adata.obs['clusters'].cat.categories)
|
| 233 |
+
ad.uns['celltype_colors']=adata.uns['clusters_colors']
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
# In[19]:
|
| 237 |
+
|
| 238 |
+
|
| 239 |
+
ov.pl.embedding(ad, basis='X_umap',
|
| 240 |
+
color=["celltype","S_score"],
|
| 241 |
+
frameon='small',cmap='RdBu_r',
|
| 242 |
+
wspace=0.5)
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
# In[ ]:
|
| 246 |
+
|
| 247 |
+
|
| 248 |
+
|
| 249 |
+
|
ovrawm/t_metatime.txt
ADDED
|
@@ -0,0 +1,177 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Celltype auto annotation with MetaTiME
|
| 5 |
+
#
|
| 6 |
+
# MetaTiME learns data-driven, interpretable, and reproducible gene programs by integrating millions of single cells from hundreds of tumor scRNA-seq data. The idea is to learn a map of single-cell space with biologically meaningful directions from large-scale data, which helps understand functional cell states and transfers knowledge to new data analysis. MetaTiME provides pretrained meta-components (MeCs) to automatically annotate fine-grained cell states and plot signature continuum for new single-cells of tumor microenvironment.
|
| 7 |
+
#
|
| 8 |
+
# Here, we integrate MetaTiME in omicverse. This tutorial demonstrates how to use [MetaTiME (original code)](https://github.com/yi-zhang/MetaTiME/blob/main/docs/notebooks/metatime_annotator.ipynb) to annotate celltype in TME
|
| 9 |
+
#
|
| 10 |
+
# Paper: [MetaTiME integrates single-cell gene expression to characterize the meta-components of the tumor immune microenvironment](https://www.nature.com/articles/s41467-023-38333-8)
|
| 11 |
+
#
|
| 12 |
+
# Code: https://github.com/yi-zhang/MetaTiME
|
| 13 |
+
#
|
| 14 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1isvjTfSFM2cy6GzHWAwbuvSjveEJijzP?usp=sharing
|
| 15 |
+
#
|
| 16 |
+
# 
|
| 17 |
+
|
| 18 |
+
# In[1]:
|
| 19 |
+
|
| 20 |
+
|
| 21 |
+
import omicverse as ov
|
| 22 |
+
ov.utils.ov_plot_set()
|
| 23 |
+
|
| 24 |
+
|
| 25 |
+
# ## Data normalize and Batch remove
|
| 26 |
+
#
|
| 27 |
+
# The sample data has multiple patients , and we can use batch correction on patients. Here, we using [scVI](https://docs.scvi-tools.org/en/stable/) to remove batch.
|
| 28 |
+
#
|
| 29 |
+
# <div class="admonition warning">
|
| 30 |
+
# <p class="admonition-title">Note</p>
|
| 31 |
+
# <p>
|
| 32 |
+
# If your data contains count matrix, we provide a wrapped function for pre-processing the data. Otherwise, if the data is already depth-normalized, log-transformed, and cells are filtered, we can skip this step.
|
| 33 |
+
# </p>
|
| 34 |
+
# </div>
|
| 35 |
+
|
| 36 |
+
# In[ ]:
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
'''
|
| 40 |
+
import scvi
|
| 41 |
+
scvi.model.SCVI.setup_anndata(adata, layer="counts", batch_key="patient")
|
| 42 |
+
vae = scvi.model.SCVI(adata, n_layers=2, n_latent=30, gene_likelihood="nb")
|
| 43 |
+
vae.train()
|
| 44 |
+
adata.obsm["X_scVI"] = vae.get_latent_representation()
|
| 45 |
+
'''
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# Example data can be obtained from figshare: https://figshare.com/ndownloader/files/41440050
|
| 49 |
+
|
| 50 |
+
# In[2]:
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
import scanpy as sc
|
| 54 |
+
adata=sc.read('TiME_adata_scvi.h5ad')
|
| 55 |
+
adata
|
| 56 |
+
|
| 57 |
+
|
| 58 |
+
# It is recommended that malignant cells are identified first and removed for best practice in cell state annotation.
|
| 59 |
+
#
|
| 60 |
+
# In the BCC data, the cluster of malignant cells are identified with `inferCNV`. We can use the pre-saved column 'isTME' to keep Tumor Microenvironment cells.
|
| 61 |
+
#
|
| 62 |
+
# These are the authors' exact words, but tests have found that the difference in annotation effect is not that great even without removing the malignant cells
|
| 63 |
+
#
|
| 64 |
+
# But I think this step is not necessary
|
| 65 |
+
|
| 66 |
+
# In[3]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
#adata = adata[adata.obs['isTME']]
|
| 70 |
+
|
| 71 |
+
|
| 72 |
+
# ## Neighborhood graph calculated
|
| 73 |
+
#
|
| 74 |
+
# We note that scVI was used earlier to remove the batch effect from the data, so we need to recalculate the neighbourhood map based on what is stored in `adata.obsm['X_scVI']`. Note that if you are not using scVI but using another method to calculate the neighbourhood map, such as `X_pca`, then you need to change `X_scVI` to `X_pca` to complete the calculation
|
| 75 |
+
#
|
| 76 |
+
# ```
|
| 77 |
+
# #Example
|
| 78 |
+
# #sc.tl.pca(adata)
|
| 79 |
+
# #sc.pp.neighbors(adata, use_rep="X_pca")
|
| 80 |
+
# ```
|
| 81 |
+
|
| 82 |
+
# In[4]:
|
| 83 |
+
|
| 84 |
+
|
| 85 |
+
sc.pp.neighbors(adata, use_rep="X_scVI")
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
# To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated.
|
| 89 |
+
|
| 90 |
+
# In[5]:
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["X_scVI"])
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
# In[6]:
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
sc.pl.embedding(
|
| 100 |
+
adata,
|
| 101 |
+
basis="X_mde",
|
| 102 |
+
color=["patient"],
|
| 103 |
+
frameon=False,
|
| 104 |
+
ncols=1,
|
| 105 |
+
)
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
# In[7]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
#adata.write_h5ad('adata_mde.h5ad',compression='gzip')
|
| 112 |
+
#adata=sc.read('adata_mde.h5ad')
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# ## MeteTiME model init
|
| 116 |
+
#
|
| 117 |
+
# Next, let's load the pre-computed MetaTiME MetaComponents (MeCs), and their functional annotation.
|
| 118 |
+
|
| 119 |
+
# In[8]:
|
| 120 |
+
|
| 121 |
+
|
| 122 |
+
TiME_object=ov.single.MetaTiME(adata,mode='table')
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
# We can over-cluster the cells which is useful for fine-grained cell state annotation.
|
| 126 |
+
#
|
| 127 |
+
# As the resolution gets larger, the number of clusters gets larger
|
| 128 |
+
|
| 129 |
+
# In[9]:
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
TiME_object.overcluster(resolution=8,clustercol = 'overcluster',)
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
# ## TME celltype predicted
|
| 136 |
+
#
|
| 137 |
+
# We using `TiME_object.predictTiME()` to predicted the latent celltype in TME.
|
| 138 |
+
#
|
| 139 |
+
# - The minor celltype will be stored in `adata.obs['MetaTiME']`
|
| 140 |
+
# - The major celltype will be stored in `adata.obs['Major_MetaTiME']`
|
| 141 |
+
|
| 142 |
+
# In[10]:
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
TiME_object.predictTiME(save_obs_name='MetaTiME')
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
# ## Visualize
|
| 149 |
+
#
|
| 150 |
+
# The original author provides a drawing function that effectively avoids overlapping labels. Here I have expanded its parameters so that it can be visualised using parameters other than X_umap
|
| 151 |
+
|
| 152 |
+
# In[13]:
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
fig,ax=TiME_object.plot(cluster_key='MetaTiME',basis='X_mde',dpi=80)
|
| 156 |
+
#fig.save
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# We can also use `sc.pl.embedding` to visualize the celltype
|
| 160 |
+
|
| 161 |
+
# In[15]:
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
sc.pl.embedding(
|
| 165 |
+
adata,
|
| 166 |
+
basis="X_mde",
|
| 167 |
+
color=["Major_MetaTiME"],
|
| 168 |
+
frameon=False,
|
| 169 |
+
ncols=1,
|
| 170 |
+
)
|
| 171 |
+
|
| 172 |
+
|
| 173 |
+
# In[ ]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
|
ovrawm/t_mofa.txt
ADDED
|
@@ -0,0 +1,184 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Multi omics analysis by MOFA
|
| 5 |
+
# MOFA is a factor analysis model that provides a general framework for the integration of multi-omic data sets in an unsupervised fashion.
|
| 6 |
+
#
|
| 7 |
+
# This tutorial focuses on how to perform mofa in multi-omics like scRNA-seq and scATAC-seq
|
| 8 |
+
#
|
| 9 |
+
# Paper: [MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02015-1)
|
| 10 |
+
#
|
| 11 |
+
# Code: https://github.com/bioFAM/mofapy2
|
| 12 |
+
#
|
| 13 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1UPGQA3BenrC-eLIGVtdKVftSnOKIwNrP?usp=sharing
|
| 14 |
+
|
| 15 |
+
# ## Part.1 MOFA Model
|
| 16 |
+
# In this part, we construct a model of mofa by scRNA-seq and scATAC-seq
|
| 17 |
+
|
| 18 |
+
# In[1]:
|
| 19 |
+
|
| 20 |
+
|
| 21 |
+
import omicverse as ov
|
| 22 |
+
rna=ov.utils.read('data/sample/rna_p_n_raw.h5ad')
|
| 23 |
+
atac=ov.utils.read('data/sample/atac_p_n_raw.h5ad')
|
| 24 |
+
|
| 25 |
+
|
| 26 |
+
# In[2]:
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
rna,atac
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
# We only need to add anndata to `ov.single.mofa` to construct the base model
|
| 33 |
+
|
| 34 |
+
# In[4]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
test_mofa=ov.single.pyMOFA(omics=[rna,atac],
|
| 38 |
+
omics_name=['RNA','ATAC'])
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
# In[ ]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
test_mofa.mofa_preprocess()
|
| 45 |
+
test_mofa.mofa_run(outfile='models/brac_rna_atac.hdf5')
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# ## Part.2 MOFA Analysis
|
| 49 |
+
# After get the model by mofa, we need to analysis the factor about different omics, we provide some method to do this
|
| 50 |
+
|
| 51 |
+
# ### load data
|
| 52 |
+
|
| 53 |
+
# In[1]:
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
import omicverse as ov
|
| 57 |
+
ov.utils.ov_plot_set()
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
# In[2]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
rna=ov.utils.read('data/sample/rna_test.h5ad')
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
# ### add value of factor to anndata
|
| 67 |
+
|
| 68 |
+
# In[3]:
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
rna=ov.single.factor_exact(rna,hdf5_path='data/sample/MOFA_POS.hdf5')
|
| 72 |
+
rna
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
# ### analysis of the correlation between factor and cell type
|
| 76 |
+
|
| 77 |
+
# In[4]:
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
ov.single.factor_correlation(adata=rna,cluster='cell_type',factor_list=[1,2,3,4,5])
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
# ### Get the gene/feature weights of different factor
|
| 84 |
+
|
| 85 |
+
# In[5]:
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
ov.single.get_weights(hdf5_path='data/sample/MOFA_POS.hdf5',view='RNA',factor=1)
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# ## Part.3 MOFA Visualize
|
| 92 |
+
#
|
| 93 |
+
# To visualize the result of mofa, we provide a series of function to do this.
|
| 94 |
+
|
| 95 |
+
# In[6]:
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
pymofa_obj=ov.single.pyMOFAART(model_path='data/sample/MOFA_POS.hdf5')
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
# We get the factor of each cell at first
|
| 102 |
+
|
| 103 |
+
# In[7]:
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
pymofa_obj.get_factors(rna)
|
| 107 |
+
rna
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
# We can also plot the varience in each View
|
| 111 |
+
|
| 112 |
+
# In[8]:
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
pymofa_obj.plot_r2()
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# In[9]:
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
pymofa_obj.get_r2()
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# ### Visualize the correlation between factor and celltype
|
| 125 |
+
|
| 126 |
+
# In[10]:
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
pymofa_obj.plot_cor(rna,'cell_type')
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# We found that factor6 is correlated to Epithelial
|
| 133 |
+
|
| 134 |
+
# In[11]:
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
pymofa_obj.plot_factor(rna,'cell_type','Epi',figsize=(3,3),
|
| 138 |
+
factor1=6,factor2=10,)
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
# In[24]:
|
| 142 |
+
|
| 143 |
+
|
| 144 |
+
import scanpy as sc
|
| 145 |
+
sc.pp.neighbors(rna)
|
| 146 |
+
sc.tl.umap(rna)
|
| 147 |
+
sc.pl.embedding(
|
| 148 |
+
rna,
|
| 149 |
+
basis="X_umap",
|
| 150 |
+
color=["factor6","cell_type"],
|
| 151 |
+
frameon=False,
|
| 152 |
+
ncols=2,
|
| 153 |
+
#palette=ov.utils.pyomic_palette(),
|
| 154 |
+
show=False,
|
| 155 |
+
cmap='Greens',
|
| 156 |
+
vmin=0,
|
| 157 |
+
)
|
| 158 |
+
#plt.savefig("figures/umap_factor6.png",dpi=300,bbox_inches = 'tight')
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
# In[12]:
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
pymofa_obj.plot_weight_gene_d1(view='RNA',factor1=6,factor2=10,)
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# In[18]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
pymofa_obj.plot_weights(view='RNA',factor=6,color='#5de25d',
|
| 171 |
+
ascending=True)
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# In[14]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
pymofa_obj.plot_top_feature_heatmap(view='RNA')
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
# In[ ]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
|
| 184 |
+
|
ovrawm/t_mofa_glue.txt
ADDED
|
@@ -0,0 +1,255 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Multi omics analysis by MOFA and GLUE
|
| 5 |
+
# MOFA is a factor analysis model that provides a general framework for the integration of multi-omic data sets in an unsupervised fashion.
|
| 6 |
+
#
|
| 7 |
+
# Most of the time, however, we did not get paired cells in the multi-omics analysis. Here, we can pair cells using GLUE, a dimensionality reduction algorithm that can integrate different histological layers, and it can efficiently merge data from different histological layers.
|
| 8 |
+
#
|
| 9 |
+
# This tutorial focuses on how to perform mofa in multi-omics using GLUE.
|
| 10 |
+
#
|
| 11 |
+
# Paper: [MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02015-1) and [Multi-omics single-cell data integration and regulatory inference with graph-linked embedding](https://www.nature.com/articles/s41587-022-01284-4)
|
| 12 |
+
#
|
| 13 |
+
# Code: https://github.com/bioFAM/mofapy2 and https://github.com/gao-lab/GLUE
|
| 14 |
+
#
|
| 15 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1zlakFf20IoBdyuOQDocwFQHu8XOVizRL?usp=sharing
|
| 16 |
+
#
|
| 17 |
+
# We used the result anndata object `rna-emb.h5ad` and `atac.emb.h5ad` from [GLUE'tutorial](https://scglue.readthedocs.io/en/latest/training.html)
|
| 18 |
+
|
| 19 |
+
# In[1]:
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
import omicverse as ov
|
| 23 |
+
ov.utils.ov_plot_set()
|
| 24 |
+
|
| 25 |
+
|
| 26 |
+
# ## Load the data
|
| 27 |
+
#
|
| 28 |
+
# We use `ov.utils.read` to read the `h5ad` files
|
| 29 |
+
|
| 30 |
+
# In[2]:
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
rna=ov.utils.read("chen_rna-emb.h5ad")
|
| 34 |
+
atac=ov.utils.read("chen_atac-emb.h5ad")
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
# ## Pair the omics
|
| 38 |
+
#
|
| 39 |
+
# Each cell in our rna and atac data has a feature vector, X_glue, based on which we can calculate the Pearson correlation coefficient to perform cell matching.
|
| 40 |
+
|
| 41 |
+
# In[3]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
pair_obj=ov.single.GLUE_pair(rna,atac)
|
| 45 |
+
pair_obj.correlation()
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# We counted the top 50 highly correlated cells in another histology layer for each cell in one of the histology layers to avoid missing data due to one cell being highly correlated with multiple cells. The default minimum threshold for high correlation is 0.9. We can obtain more paired cells by increasing the depth, but note that increasing the depth may lead to higher errors in cell matching
|
| 49 |
+
|
| 50 |
+
# In[4]:
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
res_pair=pair_obj.find_neighbor_cell(depth=20)
|
| 54 |
+
res_pair.to_csv('models/chen_pair_res.csv')
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
# We filter to get paired cells
|
| 58 |
+
|
| 59 |
+
# In[14]:
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
rna1=rna[res_pair['omic_1']]
|
| 63 |
+
atac1=atac[res_pair['omic_2']]
|
| 64 |
+
rna1.obs.index=res_pair.index
|
| 65 |
+
atac1.obs.index=res_pair.index
|
| 66 |
+
rna1,atac1
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
# We can use mudata to store the multi-omics
|
| 70 |
+
|
| 71 |
+
# In[6]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
from mudata import MuData
|
| 75 |
+
|
| 76 |
+
mdata = MuData({'rna': rna1, 'atac': atac1})
|
| 77 |
+
mdata
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
# In[7]:
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
mdata.write("chen_mu.h5mu",compression='gzip')
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# ## MOFA prepare
|
| 87 |
+
#
|
| 88 |
+
# In the MOFA analysis, we only need to use highly variable genes, for which we perform one filter
|
| 89 |
+
|
| 90 |
+
# In[22]:
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
rna1=mdata['rna']
|
| 94 |
+
rna1=rna1[:,rna1.var['highly_variable']==True]
|
| 95 |
+
atac1=mdata['atac']
|
| 96 |
+
atac1=atac1[:,atac1.var['highly_variable']==True]
|
| 97 |
+
rna1.obs.index=res_pair.index
|
| 98 |
+
atac1.obs.index=res_pair.index
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
# In[23]:
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
import random
|
| 105 |
+
random_obs_index=random.sample(list(rna1.obs.index),5000)
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
# In[25]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
from sklearn.metrics import adjusted_rand_score as ari
|
| 112 |
+
ari_random=ari(rna1[random_obs_index].obs['cell_type'], atac1[random_obs_index].obs['cell_type'])
|
| 113 |
+
ari_raw=ari(rna1.obs['cell_type'], atac1.obs['cell_type'])
|
| 114 |
+
print('raw ari:{}, random ari:{}'.format(ari_raw,ari_random))
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
# In[26]:
|
| 118 |
+
|
| 119 |
+
|
| 120 |
+
#rna1=rna1[random_obs_index]
|
| 121 |
+
#atac1=atac1[random_obs_index]
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# ## MOFA analysis
|
| 125 |
+
#
|
| 126 |
+
# In this part, we construct a model of mofa by scRNA-seq and scATAC-seq
|
| 127 |
+
|
| 128 |
+
# In[28]:
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
test_mofa=ov.single.pyMOFA(omics=[rna1,atac1],
|
| 132 |
+
omics_name=['RNA','ATAC'])
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
# In[29]:
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
test_mofa.mofa_preprocess()
|
| 139 |
+
test_mofa.mofa_run(outfile='models/chen_rna_atac.hdf5')
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
# ## MOFA Visualization
|
| 143 |
+
#
|
| 144 |
+
# In this part, we provide a series of function to visualize the result of mofa.
|
| 145 |
+
|
| 146 |
+
# In[30]:
|
| 147 |
+
|
| 148 |
+
|
| 149 |
+
pymofa_obj=ov.single.pyMOFAART(model_path='models/chen_rna_atac.hdf5')
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
# In[31]:
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
pymofa_obj.get_factors(rna1)
|
| 156 |
+
rna1
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# ### Visualize the varience of each view
|
| 160 |
+
|
| 161 |
+
# In[32]:
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
pymofa_obj.plot_r2()
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# In[33]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
pymofa_obj.get_r2()
|
| 171 |
+
|
| 172 |
+
|
| 173 |
+
# ### Visualize the correlation between factor and celltype
|
| 174 |
+
|
| 175 |
+
# In[37]:
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
pymofa_obj.plot_cor(rna1,'cell_type',figsize=(4,6))
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
# In[38]:
|
| 182 |
+
|
| 183 |
+
|
| 184 |
+
pymofa_obj.get_cor(rna1,'cell_type')
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# In[46]:
|
| 188 |
+
|
| 189 |
+
|
| 190 |
+
pymofa_obj.plot_factor(rna1,'cell_type','Ast',figsize=(3,3),
|
| 191 |
+
factor1=1,factor2=3,)
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
# ### Visualize the factor in UMAP
|
| 195 |
+
#
|
| 196 |
+
# To visualize the GLUE’s learned embeddings, we use the pymde package wrapperin scvi-tools. This is an alternative to UMAP that is GPU-accelerated.
|
| 197 |
+
#
|
| 198 |
+
# You can use `sc.tl.umap` insteaded.
|
| 199 |
+
|
| 200 |
+
# In[41]:
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
from scvi.model.utils import mde
|
| 204 |
+
import scanpy as sc
|
| 205 |
+
sc.pp.neighbors(rna1, use_rep="X_glue", metric="cosine")
|
| 206 |
+
rna1.obsm["X_mde"] = mde(rna1.obsm["X_glue"])
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
# In[47]:
|
| 210 |
+
|
| 211 |
+
|
| 212 |
+
sc.pl.embedding(
|
| 213 |
+
rna1,
|
| 214 |
+
basis="X_mde",
|
| 215 |
+
color=["factor1","factor3","cell_type"],
|
| 216 |
+
frameon=False,
|
| 217 |
+
ncols=3,
|
| 218 |
+
#palette=ov.utils.pyomic_palette(),
|
| 219 |
+
show=False,
|
| 220 |
+
cmap='Greens',
|
| 221 |
+
vmin=0,
|
| 222 |
+
)
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
# ### Weights ranked
|
| 226 |
+
# A visualization of factor weights familiar to MOFA and MOFA+ users is implemented with some modifications in `plot_weight_gene_d1`, `plot_weight_gene_d2`, and `plot_weights`.
|
| 227 |
+
|
| 228 |
+
# In[48]:
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
pymofa_obj.plot_weight_gene_d1(view='RNA',factor1=1,factor2=3,)
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
# In[50]:
|
| 235 |
+
|
| 236 |
+
|
| 237 |
+
pymofa_obj.plot_weights(view='RNA',factor=1,
|
| 238 |
+
ascending=False)
|
| 239 |
+
|
| 240 |
+
|
| 241 |
+
# ### Weights heatmap
|
| 242 |
+
#
|
| 243 |
+
# While trying to annotate factors, a global overview of top features defining them could be helpful.
|
| 244 |
+
|
| 245 |
+
# In[51]:
|
| 246 |
+
|
| 247 |
+
|
| 248 |
+
pymofa_obj.plot_top_feature_heatmap(view='RNA')
|
| 249 |
+
|
| 250 |
+
|
| 251 |
+
# In[ ]:
|
| 252 |
+
|
| 253 |
+
|
| 254 |
+
|
| 255 |
+
|
ovrawm/t_network.txt
ADDED
|
@@ -0,0 +1,88 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Protein-Protein interaction (PPI) analysis by String-db
|
| 5 |
+
# STRING is a database of known and predicted protein-protein interactions. The interactions include direct (physical) and indirect (functional) associations; they stem from computational prediction, from knowledge transfer between organisms, and from interactions aggregated from other (primary) databases.
|
| 6 |
+
#
|
| 7 |
+
# Here we produce a tutorial that use python to construct protein-protein interaction network
|
| 8 |
+
#
|
| 9 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1ReLCFA5cNNcem_WaMXYN9da7W0GN4gzl?usp=sharing
|
| 10 |
+
|
| 11 |
+
# In[1]:
|
| 12 |
+
|
| 13 |
+
|
| 14 |
+
import omicverse as ov
|
| 15 |
+
ov.utils.ov_plot_set()
|
| 16 |
+
|
| 17 |
+
|
| 18 |
+
# ## Prepare data
|
| 19 |
+
#
|
| 20 |
+
# Here we use the example data of string-db to perform the analysis
|
| 21 |
+
#
|
| 22 |
+
# FAA4 and its ten most confident interactors.
|
| 23 |
+
# FAA4 in yeast is a long chain fatty acyl-CoA synthetase; see it connected to other synthetases as well as regulators.
|
| 24 |
+
#
|
| 25 |
+
# Saccharomyces cerevisiae
|
| 26 |
+
# NCBI taxonomy Id: 4932
|
| 27 |
+
# Other names: ATCC 18824, Candida robusta, NRRL Y-12632, S. cerevisiae, Saccharomyces capensis, Saccharomyces italicus, Saccharomyces oviformis, Saccharomyces uvarum var. melibiosus, lager beer yeast, yeast
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
gene_list=['FAA4','POX1','FAT1','FAS2','FAS1','FAA1','OLE1','YJU3','TGL3','INA1','TGL5']
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# Besides, we also need to set the gene's type and color. Here, we randomly set the top 5 genes named Type1, other named Type2
|
| 36 |
+
|
| 37 |
+
# In[3]:
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
gene_type_dict=dict(zip(gene_list,['Type1']*5+['Type2']*6))
|
| 41 |
+
gene_color_dict=dict(zip(gene_list,['#F7828A']*5+['#9CCCA4']*6))
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# ## STRING interaction analysis
|
| 45 |
+
#
|
| 46 |
+
# The network API method also allows you to retrieve your STRING interaction network for one or multiple proteins in various text formats. It will tell you the combined score and all the channel specific scores for the set of proteins. You can also extend the network neighborhood by setting "add_nodes", which will add, to your network, new interaction partners in order of their confidence.
|
| 47 |
+
|
| 48 |
+
# In[7]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
G_res=ov.bulk.string_interaction(gene_list,4932)
|
| 52 |
+
G_res.head()
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
# ## STRING PPI network
|
| 56 |
+
#
|
| 57 |
+
# We also can use `ov.bulk.pyPPI` to get the PPI network of `gene_list`, we init it at first
|
| 58 |
+
|
| 59 |
+
# In[5]:
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
ppi=ov.bulk.pyPPI(gene=gene_list,
|
| 63 |
+
gene_type_dict=gene_type_dict,
|
| 64 |
+
gene_color_dict=gene_color_dict,
|
| 65 |
+
species=4932)
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
# Then we connect to string-db to calculate the protein-protein interaction
|
| 69 |
+
|
| 70 |
+
# In[8]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
ppi.interaction_analysis()
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
# We provided a very simple function to plot the network, you can refer the `ov.utils.plot_network` to find out the parameter
|
| 77 |
+
|
| 78 |
+
# In[9]:
|
| 79 |
+
|
| 80 |
+
|
| 81 |
+
ppi.plot_network()
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
# In[ ]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
|
ovrawm/t_nocd.txt
ADDED
|
@@ -0,0 +1,112 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Overlapping Celltype annotation with GNN
|
| 5 |
+
# Droplet based single cell transcriptomics has recently enabled parallel screening of tens of thousands of single cells. Clustering methods that scale for such high dimensional data without compromising accuracy are scarce.
|
| 6 |
+
#
|
| 7 |
+
# This tutorial focuses on how to cluster the cell with overlapping and identify the cell with multi-fate
|
| 8 |
+
#
|
| 9 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1l7iHdVmTQcv9YmpIjhK_UzLuHbJ1Jv9E?usp=sharing
|
| 10 |
+
#
|
| 11 |
+
# <div class="admonition warning">
|
| 12 |
+
# <p class="admonition-title">Warning</p>
|
| 13 |
+
# <p>
|
| 14 |
+
# NOCD's development is still in progress. The current version may not fully reproduce the original implementation’s results.
|
| 15 |
+
# </p>
|
| 16 |
+
# </div>
|
| 17 |
+
|
| 18 |
+
# ## Part.1 Data preprocess
|
| 19 |
+
#
|
| 20 |
+
# In this part, we perform preliminary processing of the data, such as normalization and logarithmization, in order to make the data more interpretable
|
| 21 |
+
|
| 22 |
+
# In[1]:
|
| 23 |
+
|
| 24 |
+
|
| 25 |
+
import omicverse as ov
|
| 26 |
+
import anndata
|
| 27 |
+
import scanpy as sc
|
| 28 |
+
import matplotlib.pyplot as plt
|
| 29 |
+
import numpy as np
|
| 30 |
+
import pandas as pd
|
| 31 |
+
get_ipython().run_line_magic('matplotlib', 'inline')
|
| 32 |
+
|
| 33 |
+
|
| 34 |
+
# In[2]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
#param for visualization
|
| 38 |
+
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
|
| 39 |
+
sc.settings.set_figure_params(dpi=80, facecolor='white')
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
# In[3]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
from matplotlib.colors import LinearSegmentedColormap
|
| 46 |
+
sc_color=['#7CBB5F','#368650','#A499CC','#5E4D9A','#78C2ED','#866017', '#9F987F','#E0DFED',
|
| 47 |
+
'#EF7B77', '#279AD7','#F0EEF0', '#1F577B', '#A56BA7', '#E0A7C8', '#E069A6', '#941456', '#FCBC10',
|
| 48 |
+
'#EAEFC5', '#01A0A7', '#75C8CC', '#F0D7BC', '#D5B26C', '#D5DA48', '#B6B812', '#9DC3C3', '#A89C92', '#FEE00C', '#FEF2A1']
|
| 49 |
+
sc_color_cmap = LinearSegmentedColormap.from_list('Custom', sc_color, len(sc_color))
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
# In[4]:
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
adata = anndata.read('sample/rna.h5ad')
|
| 56 |
+
adata
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
# In[5]:
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
adata=ov.single.scanpy_lazy(adata)
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
# ## Part.2 Overlapping Community Detection
|
| 66 |
+
# In this part, we perform a graph neural network (GNN) basedmodel for overlapping community detection in scRNA-seq.
|
| 67 |
+
#
|
| 68 |
+
# 
|
| 69 |
+
|
| 70 |
+
# In[6]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
scbrca=ov.single.scnocd(adata)
|
| 74 |
+
scbrca.matrix_transform()
|
| 75 |
+
scbrca.matrix_normalize()
|
| 76 |
+
scbrca.GNN_configure()
|
| 77 |
+
scbrca.GNN_preprocess()
|
| 78 |
+
scbrca.GNN_model()
|
| 79 |
+
scbrca.GNN_result()
|
| 80 |
+
scbrca.GNN_plot()
|
| 81 |
+
#scbrca.calculate_nocd()
|
| 82 |
+
scbrca.cal_nocd()
|
| 83 |
+
|
| 84 |
+
|
| 85 |
+
# In[8]:
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
scbrca.calculate_nocd()
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# ## Part.3 Visualization
|
| 92 |
+
# In this part, we visualized the overlapping and non-overlapping cell.
|
| 93 |
+
|
| 94 |
+
# In[9]:
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
sc.pl.umap(scbrca.adata, color=['leiden','nocd'],wspace=0.4,palette=sc_color)
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# zero means the cell related to overlap
|
| 101 |
+
|
| 102 |
+
# In[10]:
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
sc.pl.umap(scbrca.adata, color=['leiden','nocd_n'],wspace=0.4,palette=sc_color)
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
# In[ ]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
|
ovrawm/t_preprocess.txt
ADDED
|
@@ -0,0 +1,421 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Preprocessing the data of scRNA-seq with omicverse
|
| 5 |
+
#
|
| 6 |
+
# The count table, a numeric matrix of genes × cells, is the basic input data structure in the analysis of single-cell RNA-sequencing data. A common preprocessing step is to adjust the counts for variable sampling efficiency and to transform them so that the variance is similar across the dynamic range.
|
| 7 |
+
#
|
| 8 |
+
# Suitable methods to preprocess the scRNA-seq is important. Here, we introduce some preprocessing step to help researchers can perform downstream analysis easyier.
|
| 9 |
+
#
|
| 10 |
+
# User can compare our tutorial with [scanpy'tutorial](https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html) to learn how to use omicverse well
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1DXLSls_ppgJmAaZTUvqazNC_E7EDCxUe?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
import scanpy as sc
|
| 19 |
+
ov.ov_plot_set()
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
# The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics ([here](http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz) from this [webpage](https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k)). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
|
| 23 |
+
|
| 24 |
+
# In[2]:
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# !mkdir data
|
| 28 |
+
# !wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 29 |
+
# !cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 30 |
+
# !mkdir write
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
# In[3]:
|
| 34 |
+
|
| 35 |
+
|
| 36 |
+
adata = sc.read_10x_mtx(
|
| 37 |
+
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
|
| 38 |
+
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
|
| 39 |
+
cache=True) # write a cache file for faster subsequent reading
|
| 40 |
+
adata
|
| 41 |
+
|
| 42 |
+
|
| 43 |
+
# In[4]:
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
adata.var_names_make_unique()
|
| 47 |
+
adata.obs_names_make_unique()
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
# ## Preprocessing
|
| 51 |
+
#
|
| 52 |
+
# ### Quantity control
|
| 53 |
+
#
|
| 54 |
+
# For single-cell data, we require quality control prior to analysis, including the removal of cells containing double cells, low-expressing cells, and low-expressing genes. In addition to this, we need to filter based on mitochondrial gene ratios, number of transcripts, number of genes expressed per cell, cellular Complexity, etc. For a detailed description of the different QCs please see the document: https://hbctraining.github.io/scRNA-seq/lessons/04_SC_quality_control.html
|
| 55 |
+
|
| 56 |
+
# In[5]:
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
adata=ov.pp.qc(adata,
|
| 60 |
+
tresh={'mito_perc': 0.05, 'nUMIs': 500, 'detected_genes': 250})
|
| 61 |
+
adata
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# ### High variable Gene Detection
|
| 65 |
+
#
|
| 66 |
+
# Here we try to use Pearson's method to calculate highly variable genes. This is the method that is proposed to be superior to ordinary normalisation. See [Article](https://www.nature.com/articles/s41592-023-01814-1#MOESM3) in *Nature Method* for details.
|
| 67 |
+
#
|
| 68 |
+
|
| 69 |
+
# Sometimes we need to recover the original counts for some single-cell calculations, but storing them in the layer layer may result in missing data, so we provide two functions here, a store function and a release function, to save the original data.
|
| 70 |
+
#
|
| 71 |
+
# We set `layers=counts`, the counts will be stored in `adata.uns['layers_counts']`
|
| 72 |
+
|
| 73 |
+
# In[6]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
ov.utils.store_layers(adata,layers='counts')
|
| 77 |
+
adata
|
| 78 |
+
|
| 79 |
+
|
| 80 |
+
# normalize|HVGs:We use | to control the preprocessing step, | before for the normalisation step, either `shiftlog` or `pearson`, and | after for the highly variable gene calculation step, either `pearson` or `seurat`. Our default is `shiftlog|pearson`.
|
| 81 |
+
#
|
| 82 |
+
# - if you use `mode`=`shiftlog|pearson` you need to set `target_sum=50*1e4`, more people like to se `target_sum=1e4`, we test the result think 50*1e4 will be better
|
| 83 |
+
# - if you use `mode`=`pearson|pearson`, you don't need to set `target_sum`
|
| 84 |
+
#
|
| 85 |
+
# <div class="admonition warning">
|
| 86 |
+
# <p class="admonition-title">Note</p>
|
| 87 |
+
# <p>
|
| 88 |
+
# if the version of `omicverse` lower than `1.4.13`, the mode can only be set between `scanpy` and `pearson`.
|
| 89 |
+
# </p>
|
| 90 |
+
# </div>
|
| 91 |
+
#
|
| 92 |
+
|
| 93 |
+
# In[7]:
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 97 |
+
adata
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# Set the .raw attribute of the AnnData object to the normalized and logarithmized raw gene expression for later use in differential testing and visualizations of gene expression. This simply freezes the state of the AnnData object.
|
| 101 |
+
|
| 102 |
+
# In[8]:
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
adata.raw = adata
|
| 106 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 107 |
+
adata
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
# We find that the adata.X matrix is normalized at this point, including the data in raw, but we want to get the unnormalized data, so we can use the retrieve function `ov.utils.retrieve_layers`
|
| 111 |
+
|
| 112 |
+
# In[9]:
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
adata_counts=adata.copy()
|
| 116 |
+
ov.utils.retrieve_layers(adata_counts,layers='counts')
|
| 117 |
+
print('normalize adata:',adata.X.max())
|
| 118 |
+
print('raw count adata:',adata_counts.X.max())
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# In[10]:
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
adata_counts
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
# If we wish to recover the original count matrix at the whole gene level, we can try the following code
|
| 128 |
+
|
| 129 |
+
# In[11]:
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
adata_counts=adata.raw.to_adata().copy()
|
| 133 |
+
ov.utils.retrieve_layers(adata_counts,layers='counts')
|
| 134 |
+
print('normalize adata:',adata.X.max())
|
| 135 |
+
print('raw count adata:',adata_counts.X.max())
|
| 136 |
+
adata_counts
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
# ## Principal component analysis
|
| 140 |
+
#
|
| 141 |
+
# In contrast to scanpy, we do not directly scale the variance of the original expression matrix, but store the results of the variance scaling in the layer, due to the fact that scale may cause changes in the data distribution, and we have not found scale to be meaningful in any scenario other than a principal component analysis
|
| 142 |
+
|
| 143 |
+
# In[12]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
ov.pp.scale(adata)
|
| 147 |
+
adata
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
# If you want to perform pca in normlog layer, you can set `layer`=`normlog`, but we think scaled is necessary in PCA.
|
| 151 |
+
|
| 152 |
+
# In[13]:
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 156 |
+
adata
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# In[14]:
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
adata.obsm['X_pca']=adata.obsm['scaled|original|X_pca']
|
| 163 |
+
ov.utils.embedding(adata,
|
| 164 |
+
basis='X_pca',
|
| 165 |
+
color='CST3',
|
| 166 |
+
frameon='small')
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
# ## Embedding the neighborhood graph
|
| 170 |
+
#
|
| 171 |
+
# We suggest embedding the graph in two dimensions using UMAP (McInnes et al., 2018), see below. It is potentially more faithful to the global connectivity of the manifold than tSNE, i.e., it better preserves trajectories. In some ocassions, you might still observe disconnected clusters and similar connectivity violations. They can usually be remedied by running:
|
| 172 |
+
|
| 173 |
+
# In[15]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 177 |
+
use_rep='scaled|original|X_pca')
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
# To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated.
|
| 181 |
+
|
| 182 |
+
# In[16]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 186 |
+
adata
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# In[17]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
ov.utils.embedding(adata,
|
| 193 |
+
basis='X_mde',
|
| 194 |
+
color='CST3',
|
| 195 |
+
frameon='small')
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
# You also can use `umap` to visualize the neighborhood graph
|
| 199 |
+
|
| 200 |
+
# In[18]:
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
sc.tl.umap(adata)
|
| 204 |
+
|
| 205 |
+
|
| 206 |
+
# In[19]:
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
ov.utils.embedding(adata,
|
| 210 |
+
basis='X_umap',
|
| 211 |
+
color='CST3',
|
| 212 |
+
frameon='small')
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
# ## Clustering the neighborhood graph
|
| 216 |
+
#
|
| 217 |
+
# As with Seurat and many other frameworks, we recommend the Leiden graph-clustering method (community detection based on optimizing modularity) by Traag *et al.* (2018). Note that Leiden clustering directly clusters the neighborhood graph of cells, which we already computed in the previous section.
|
| 218 |
+
|
| 219 |
+
# In[20]:
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
sc.tl.leiden(adata)
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
# We redesigned the visualisation of embedding to distinguish it from scanpy's embedding by adding the parameter `fraemon='small'`, which causes the axes to be scaled with the colourbar
|
| 226 |
+
|
| 227 |
+
# In[21]:
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
ov.utils.embedding(adata,
|
| 231 |
+
basis='X_mde',
|
| 232 |
+
color=['leiden', 'CST3', 'NKG7'],
|
| 233 |
+
frameon='small')
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
# We also provide a boundary visualisation function `ov.utils.plot_ConvexHull` to visualise specific clusters.
|
| 237 |
+
#
|
| 238 |
+
# Arguments:
|
| 239 |
+
# - color: if None will use the color of clusters
|
| 240 |
+
# - alpha: default is 0.2
|
| 241 |
+
|
| 242 |
+
# In[23]:
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
import matplotlib.pyplot as plt
|
| 246 |
+
fig,ax=plt.subplots( figsize = (4,4))
|
| 247 |
+
|
| 248 |
+
ov.utils.embedding(adata,
|
| 249 |
+
basis='X_mde',
|
| 250 |
+
color=['leiden'],
|
| 251 |
+
frameon='small',
|
| 252 |
+
show=False,
|
| 253 |
+
ax=ax)
|
| 254 |
+
|
| 255 |
+
ov.utils.plot_ConvexHull(adata,
|
| 256 |
+
basis='X_mde',
|
| 257 |
+
cluster_key='leiden',
|
| 258 |
+
hull_cluster='0',
|
| 259 |
+
ax=ax)
|
| 260 |
+
|
| 261 |
+
|
| 262 |
+
# If you have too many labels, e.g. too many cell types, and you are concerned about cell overlap, then consider trying the `ov.utils.gen_mpl_labels` function, which improves text overlap.
|
| 263 |
+
# In addition, we make use of the `patheffects` function, which makes our text have outlines
|
| 264 |
+
#
|
| 265 |
+
# - adjust_kwargs: it could be found in package `adjusttext`
|
| 266 |
+
# - text_kwargs: it could be found in class `plt.texts`
|
| 267 |
+
|
| 268 |
+
# In[67]:
|
| 269 |
+
|
| 270 |
+
|
| 271 |
+
from matplotlib import patheffects
|
| 272 |
+
import matplotlib.pyplot as plt
|
| 273 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 274 |
+
|
| 275 |
+
ov.utils.embedding(adata,
|
| 276 |
+
basis='X_mde',
|
| 277 |
+
color=['leiden'],
|
| 278 |
+
show=False, legend_loc=None, add_outline=False,
|
| 279 |
+
frameon='small',legend_fontoutline=2,ax=ax
|
| 280 |
+
)
|
| 281 |
+
|
| 282 |
+
ov.utils.gen_mpl_labels(
|
| 283 |
+
adata,
|
| 284 |
+
'leiden',
|
| 285 |
+
exclude=("None",),
|
| 286 |
+
basis='X_mde',
|
| 287 |
+
ax=ax,
|
| 288 |
+
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
|
| 289 |
+
text_kwargs=dict(fontsize= 12 ,weight='bold',
|
| 290 |
+
path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),
|
| 291 |
+
)
|
| 292 |
+
|
| 293 |
+
|
| 294 |
+
# In[47]:
|
| 295 |
+
|
| 296 |
+
|
| 297 |
+
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
|
| 298 |
+
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
|
| 299 |
+
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
|
| 300 |
+
|
| 301 |
+
|
| 302 |
+
# In[48]:
|
| 303 |
+
|
| 304 |
+
|
| 305 |
+
sc.pl.dotplot(adata, marker_genes, groupby='leiden',
|
| 306 |
+
standard_scale='var');
|
| 307 |
+
|
| 308 |
+
|
| 309 |
+
# ## Finding marker genes
|
| 310 |
+
#
|
| 311 |
+
# Let us compute a ranking for the highly differential genes in each cluster. For this, by default, the .raw attribute of AnnData is used in case it has been initialized before. The simplest and fastest method to do so is the t-test.
|
| 312 |
+
|
| 313 |
+
# In[49]:
|
| 314 |
+
|
| 315 |
+
|
| 316 |
+
sc.tl.dendrogram(adata,'leiden',use_rep='scaled|original|X_pca')
|
| 317 |
+
sc.tl.rank_genes_groups(adata, 'leiden', use_rep='scaled|original|X_pca',
|
| 318 |
+
method='t-test',use_raw=False,key_added='leiden_ttest')
|
| 319 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 320 |
+
cmap='Spectral_r',key='leiden_ttest',
|
| 321 |
+
standard_scale='var',n_genes=3)
|
| 322 |
+
|
| 323 |
+
|
| 324 |
+
# cosg is also considered to be a better algorithm for finding marker genes. Here, omicverse provides the calculation of cosg
|
| 325 |
+
#
|
| 326 |
+
# Paper: [Accurate and fast cell marker gene identification with COSG](https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab579/6511197?redirectedFrom=fulltext)
|
| 327 |
+
#
|
| 328 |
+
# Code: https://github.com/genecell/COSG
|
| 329 |
+
#
|
| 330 |
+
|
| 331 |
+
# In[50]:
|
| 332 |
+
|
| 333 |
+
|
| 334 |
+
sc.tl.rank_genes_groups(adata, groupby='leiden',
|
| 335 |
+
method='t-test',use_rep='scaled|original|X_pca',)
|
| 336 |
+
ov.single.cosg(adata, key_added='leiden_cosg', groupby='leiden')
|
| 337 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 338 |
+
cmap='Spectral_r',key='leiden_cosg',
|
| 339 |
+
standard_scale='var',n_genes=3)
|
| 340 |
+
|
| 341 |
+
|
| 342 |
+
# ## Other plotting
|
| 343 |
+
#
|
| 344 |
+
# Next, let's try another chart, which we call the Stacked Volcano Chart. We need to prepare two dictionaries, a `data_dict` and a `color_dict`, both of which have the same key requirements.
|
| 345 |
+
#
|
| 346 |
+
# For `data_dict`. we require the contents within each key to be a DataFrame containing ['names','logfoldchanges','pvals_adj'], where names stands for gene names, logfoldchanges stands for differential expression multiplicity, pvals_adj stands for significance p-value
|
| 347 |
+
#
|
| 348 |
+
|
| 349 |
+
# In[51]:
|
| 350 |
+
|
| 351 |
+
|
| 352 |
+
data_dict={}
|
| 353 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 354 |
+
data_dict[i]=sc.get.rank_genes_groups_df(adata, group=i, key='leiden_ttest',
|
| 355 |
+
pval_cutoff=None,log2fc_min=None)
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
# In[65]:
|
| 359 |
+
|
| 360 |
+
|
| 361 |
+
data_dict.keys()
|
| 362 |
+
|
| 363 |
+
|
| 364 |
+
# In[64]:
|
| 365 |
+
|
| 366 |
+
|
| 367 |
+
data_dict[i].head()
|
| 368 |
+
|
| 369 |
+
|
| 370 |
+
# For `color_dict`, we require that the colour to be displayed for the current key is stored within each key.`
|
| 371 |
+
|
| 372 |
+
# In[63]:
|
| 373 |
+
|
| 374 |
+
|
| 375 |
+
type_color_dict=dict(zip(adata.obs['leiden'].cat.categories,
|
| 376 |
+
adata.uns['leiden_colors']))
|
| 377 |
+
type_color_dict
|
| 378 |
+
|
| 379 |
+
|
| 380 |
+
# There are a number of parameters available here for us to customise the settings. Note that when drawing stacking_vol with omicverse version less than 1.4.13, there is a bug that the vertical coordinate is constant at [-15,15], so we have added some code in this tutorial for visualisation.
|
| 381 |
+
#
|
| 382 |
+
# - data_dict: dict, in each key, there is a dataframe with columns of ['logfoldchanges','pvals_adj','names']
|
| 383 |
+
# - color_dict: dict, in each key, there is a color for each omic
|
| 384 |
+
# - pval_threshold: float, pvalue threshold for significant genes
|
| 385 |
+
# - log2fc_threshold: float, log2fc threshold for significant genes
|
| 386 |
+
# - figsize: tuple, figure size
|
| 387 |
+
# - sig_color: str, color for significant genes
|
| 388 |
+
# - normal_color: str, color for non-significant genes
|
| 389 |
+
# - plot_genes_num: int, number of genes to plot
|
| 390 |
+
# - plot_genes_fontsize: int, fontsize for gene names
|
| 391 |
+
# - plot_genes_weight: str, weight for gene names
|
| 392 |
+
|
| 393 |
+
# In[62]:
|
| 394 |
+
|
| 395 |
+
|
| 396 |
+
fig,axes=ov.utils.stacking_vol(data_dict,type_color_dict,
|
| 397 |
+
pval_threshold=0.01,
|
| 398 |
+
log2fc_threshold=2,
|
| 399 |
+
figsize=(8,4),
|
| 400 |
+
sig_color='#a51616',
|
| 401 |
+
normal_color='#c7c7c7',
|
| 402 |
+
plot_genes_num=2,
|
| 403 |
+
plot_genes_fontsize=6,
|
| 404 |
+
plot_genes_weight='bold',
|
| 405 |
+
)
|
| 406 |
+
|
| 407 |
+
#The following code will be removed in future
|
| 408 |
+
y_min,y_max=0,0
|
| 409 |
+
for i in data_dict.keys():
|
| 410 |
+
y_min=min(y_min,data_dict[i]['logfoldchanges'].min())
|
| 411 |
+
y_max=max(y_max,data_dict[i]['logfoldchanges'].max())
|
| 412 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 413 |
+
axes[i].set_ylim(y_min,y_max)
|
| 414 |
+
plt.suptitle('Stacking_vol',fontsize=12)
|
| 415 |
+
|
| 416 |
+
|
| 417 |
+
# In[ ]:
|
| 418 |
+
|
| 419 |
+
|
| 420 |
+
|
| 421 |
+
|
ovrawm/t_preprocess_cpu.txt
ADDED
|
@@ -0,0 +1,404 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Preprocessing the data of scRNA-seq with omicverse
|
| 5 |
+
#
|
| 6 |
+
# The count table, a numeric matrix of genes × cells, is the basic input data structure in the analysis of single-cell RNA-sequencing data. A common preprocessing step is to adjust the counts for variable sampling efficiency and to transform them so that the variance is similar across the dynamic range.
|
| 7 |
+
#
|
| 8 |
+
# Suitable methods to preprocess the scRNA-seq is important. Here, we introduce some preprocessing step to help researchers can perform downstream analysis easyier.
|
| 9 |
+
#
|
| 10 |
+
# User can compare our tutorial with [scanpy'tutorial](https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html) to learn how to use omicverse well
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1DXLSls_ppgJmAaZTUvqazNC_E7EDCxUe?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import scanpy as sc
|
| 18 |
+
import omicverse as ov
|
| 19 |
+
ov.plot_set()
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
# The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics ([here](http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz) from this [webpage](https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k)). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
|
| 23 |
+
|
| 24 |
+
# In[ ]:
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# !mkdir data
|
| 28 |
+
get_ipython().system('wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz')
|
| 29 |
+
get_ipython().system('cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz')
|
| 30 |
+
# !mkdir write
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
# In[2]:
|
| 34 |
+
|
| 35 |
+
|
| 36 |
+
adata = sc.read_10x_mtx(
|
| 37 |
+
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
|
| 38 |
+
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
|
| 39 |
+
cache=True) # write a cache file for faster subsequent reading
|
| 40 |
+
adata
|
| 41 |
+
|
| 42 |
+
|
| 43 |
+
# In[3]:
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
adata.var_names_make_unique()
|
| 47 |
+
adata.obs_names_make_unique()
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
# ## Preprocessing
|
| 51 |
+
#
|
| 52 |
+
# ### Quantity control
|
| 53 |
+
#
|
| 54 |
+
# For single-cell data, we require quality control prior to analysis, including the removal of cells containing double cells, low-expressing cells, and low-expressing genes. In addition to this, we need to filter based on mitochondrial gene ratios, number of transcripts, number of genes expressed per cell, cellular Complexity, etc. For a detailed description of the different QCs please see the document: https://hbctraining.github.io/scRNA-seq/lessons/04_SC_quality_control.html
|
| 55 |
+
#
|
| 56 |
+
# <div class="admonition warning">
|
| 57 |
+
# <p class="admonition-title">Note</p>
|
| 58 |
+
# <p>
|
| 59 |
+
# if the version of `omicverse` larger than `1.6.4`, the `doublets_method` can be set between `scrublet` and `sccomposite`.
|
| 60 |
+
# </p>
|
| 61 |
+
# </div>
|
| 62 |
+
#
|
| 63 |
+
# COMPOSITE (COMpound POiSson multIplet deTEction model) is a computational tool for multiplet detection in both single-cell single-omics and multiomics settings. It has been implemented as an automated pipeline and is available as both a cloud-based application with a user-friendly interface and a Python package.
|
| 64 |
+
#
|
| 65 |
+
# Hu, H., Wang, X., Feng, S. et al. A unified model-based framework for doublet or multiplet detection in single-cell multiomics data. Nat Commun 15, 5562 (2024). https://doi.org/10.1038/s41467-024-49448-x
|
| 66 |
+
|
| 67 |
+
# In[4]:
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.qc(adata,\n tresh={'mito_perc': 0.2, 'nUMIs': 500, 'detected_genes': 250},\n doublets_method='sccomposite',\n batch_key=None)\nadata\n")
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# ### High variable Gene Detection
|
| 74 |
+
#
|
| 75 |
+
# Here we try to use Pearson's method to calculate highly variable genes. This is the method that is proposed to be superior to ordinary normalisation. See [Article](https://www.nature.com/articles/s41592-023-01814-1#MOESM3) in *Nature Method* for details.
|
| 76 |
+
#
|
| 77 |
+
|
| 78 |
+
# normalize|HVGs:We use | to control the preprocessing step, | before for the normalisation step, either `shiftlog` or `pearson`, and | after for the highly variable gene calculation step, either `pearson` or `seurat`. Our default is `shiftlog|pearson`.
|
| 79 |
+
#
|
| 80 |
+
# - if you use `mode`=`shiftlog|pearson` you need to set `target_sum=50*1e4`, more people like to se `target_sum=1e4`, we test the result think 50*1e4 will be better
|
| 81 |
+
# - if you use `mode`=`pearson|pearson`, you don't need to set `target_sum`
|
| 82 |
+
#
|
| 83 |
+
# <div class="admonition warning">
|
| 84 |
+
# <p class="admonition-title">Note</p>
|
| 85 |
+
# <p>
|
| 86 |
+
# if the version of `omicverse` lower than `1.4.13`, the mode can only be set between `scanpy` and `pearson`.
|
| 87 |
+
# </p>
|
| 88 |
+
# </div>
|
| 89 |
+
#
|
| 90 |
+
|
| 91 |
+
# In[5]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)\nadata\n")
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# Set the .raw attribute of the AnnData object to the normalized and logarithmized raw gene expression for later use in differential testing and visualizations of gene expression. This simply freezes the state of the AnnData object.
|
| 98 |
+
|
| 99 |
+
# In[6]:
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
get_ipython().run_cell_magic('time', '', 'adata.raw = adata\nadata = adata[:, adata.var.highly_variable_features]\nadata\n')
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
# ## Principal component analysis
|
| 106 |
+
#
|
| 107 |
+
# In contrast to scanpy, we do not directly scale the variance of the original expression matrix, but store the results of the variance scaling in the layer, due to the fact that scale may cause changes in the data distribution, and we have not found scale to be meaningful in any scenario other than a principal component analysis
|
| 108 |
+
|
| 109 |
+
# In[7]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
get_ipython().run_cell_magic('time', '', 'ov.pp.scale(adata)\nadata\n')
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# If you want to perform pca in normlog layer, you can set `layer`=`normlog`, but we think scaled is necessary in PCA.
|
| 116 |
+
|
| 117 |
+
# In[8]:
|
| 118 |
+
|
| 119 |
+
|
| 120 |
+
get_ipython().run_cell_magic('time', '', "ov.pp.pca(adata,layer='scaled',n_pcs=50)\nadata\n")
|
| 121 |
+
|
| 122 |
+
|
| 123 |
+
# In[9]:
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
adata.obsm['X_pca']=adata.obsm['scaled|original|X_pca']
|
| 127 |
+
ov.pl.embedding(adata,
|
| 128 |
+
basis='X_pca',
|
| 129 |
+
color='CST3',
|
| 130 |
+
frameon='small')
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
# ## Embedding the neighborhood graph
|
| 134 |
+
#
|
| 135 |
+
# We suggest embedding the graph in two dimensions using UMAP (McInnes et al., 2018), see below. It is potentially more faithful to the global connectivity of the manifold than tSNE, i.e., it better preserves trajectories. In some ocassions, you might still observe disconnected clusters and similar connectivity violations. They can usually be remedied by running:
|
| 136 |
+
|
| 137 |
+
# In[10]:
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
get_ipython().run_cell_magic('time', '', "ov.pp.neighbors(adata, n_neighbors=15, n_pcs=50,\n use_rep='scaled|original|X_pca')\n")
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
# You also can use `umap` to visualize the neighborhood graph
|
| 144 |
+
|
| 145 |
+
# In[11]:
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
get_ipython().run_cell_magic('time', '', 'ov.pp.umap(adata)\n')
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
# In[12]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
ov.pl.embedding(adata,
|
| 155 |
+
basis='X_umap',
|
| 156 |
+
color='CST3',
|
| 157 |
+
frameon='small')
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
# To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated.
|
| 161 |
+
|
| 162 |
+
# In[13]:
|
| 163 |
+
|
| 164 |
+
|
| 165 |
+
ov.pp.mde(adata,embedding_dim=2,n_neighbors=15, basis='X_mde',
|
| 166 |
+
n_pcs=50, use_rep='scaled|original|X_pca',)
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
# In[14]:
|
| 170 |
+
|
| 171 |
+
|
| 172 |
+
ov.pl.embedding(adata,
|
| 173 |
+
basis='X_mde',
|
| 174 |
+
color='CST3',
|
| 175 |
+
frameon='small')
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
# ## Score cell cyle
|
| 179 |
+
#
|
| 180 |
+
# In OmicVerse, we store both G1M/S and G2M genes into the function (both human and mouse), so you can run cell cycle analysis without having to manually enter cycle genes!
|
| 181 |
+
|
| 182 |
+
# In[19]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
adata_raw=adata.raw.to_adata()
|
| 186 |
+
ov.pp.score_genes_cell_cycle(adata_raw,species='human')
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# In[21]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
ov.pl.embedding(adata_raw,
|
| 193 |
+
basis='X_mde',
|
| 194 |
+
color='phase',
|
| 195 |
+
frameon='small')
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
# ## Clustering the neighborhood graph
|
| 199 |
+
#
|
| 200 |
+
# As with Seurat and many other frameworks, we recommend the Leiden graph-clustering method (community detection based on optimizing modularity) by Traag *et al.* (2018). Note that Leiden clustering directly clusters the neighborhood graph of cells, which we already computed in the previous section.
|
| 201 |
+
|
| 202 |
+
# In[22]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
ov.pp.leiden(adata,resolution=1)
|
| 206 |
+
|
| 207 |
+
|
| 208 |
+
# We redesigned the visualisation of embedding to distinguish it from scanpy's embedding by adding the parameter `fraemon='small'`, which causes the axes to be scaled with the colourbar
|
| 209 |
+
|
| 210 |
+
# In[23]:
|
| 211 |
+
|
| 212 |
+
|
| 213 |
+
ov.pl.embedding(adata,
|
| 214 |
+
basis='X_mde',
|
| 215 |
+
color=['leiden', 'CST3', 'NKG7'],
|
| 216 |
+
frameon='small')
|
| 217 |
+
|
| 218 |
+
|
| 219 |
+
# We also provide a boundary visualisation function `ov.utils.plot_ConvexHull` to visualise specific clusters.
|
| 220 |
+
#
|
| 221 |
+
# Arguments:
|
| 222 |
+
# - color: if None will use the color of clusters
|
| 223 |
+
# - alpha: default is 0.2
|
| 224 |
+
|
| 225 |
+
# In[24]:
|
| 226 |
+
|
| 227 |
+
|
| 228 |
+
import matplotlib.pyplot as plt
|
| 229 |
+
fig,ax=plt.subplots( figsize = (4,4))
|
| 230 |
+
|
| 231 |
+
ov.pl.embedding(adata,
|
| 232 |
+
basis='X_mde',
|
| 233 |
+
color=['leiden'],
|
| 234 |
+
frameon='small',
|
| 235 |
+
show=False,
|
| 236 |
+
ax=ax)
|
| 237 |
+
|
| 238 |
+
ov.pl.ConvexHull(adata,
|
| 239 |
+
basis='X_mde',
|
| 240 |
+
cluster_key='leiden',
|
| 241 |
+
hull_cluster='0',
|
| 242 |
+
ax=ax)
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
# If you have too many labels, e.g. too many cell types, and you are concerned about cell overlap, then consider trying the `ov.utils.gen_mpl_labels` function, which improves text overlap.
|
| 246 |
+
# In addition, we make use of the `patheffects` function, which makes our text have outlines
|
| 247 |
+
#
|
| 248 |
+
# - adjust_kwargs: it could be found in package `adjusttext`
|
| 249 |
+
# - text_kwargs: it could be found in class `plt.texts`
|
| 250 |
+
|
| 251 |
+
# In[25]:
|
| 252 |
+
|
| 253 |
+
|
| 254 |
+
from matplotlib import patheffects
|
| 255 |
+
import matplotlib.pyplot as plt
|
| 256 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 257 |
+
|
| 258 |
+
ov.pl.embedding(adata,
|
| 259 |
+
basis='X_mde',
|
| 260 |
+
color=['leiden'],
|
| 261 |
+
show=False, legend_loc=None, add_outline=False,
|
| 262 |
+
frameon='small',legend_fontoutline=2,ax=ax
|
| 263 |
+
)
|
| 264 |
+
|
| 265 |
+
ov.utils.gen_mpl_labels(
|
| 266 |
+
adata,
|
| 267 |
+
'leiden',
|
| 268 |
+
exclude=("None",),
|
| 269 |
+
basis='X_mde',
|
| 270 |
+
ax=ax,
|
| 271 |
+
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
|
| 272 |
+
text_kwargs=dict(fontsize= 12 ,weight='bold',
|
| 273 |
+
path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),
|
| 274 |
+
)
|
| 275 |
+
|
| 276 |
+
|
| 277 |
+
# In[26]:
|
| 278 |
+
|
| 279 |
+
|
| 280 |
+
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
|
| 281 |
+
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
|
| 282 |
+
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
|
| 283 |
+
|
| 284 |
+
|
| 285 |
+
# In[27]:
|
| 286 |
+
|
| 287 |
+
|
| 288 |
+
sc.pl.dotplot(adata, marker_genes, groupby='leiden',
|
| 289 |
+
standard_scale='var');
|
| 290 |
+
|
| 291 |
+
|
| 292 |
+
# ## Finding marker genes
|
| 293 |
+
#
|
| 294 |
+
# Let us compute a ranking for the highly differential genes in each cluster. For this, by default, the .raw attribute of AnnData is used in case it has been initialized before. The simplest and fastest method to do so is the t-test.
|
| 295 |
+
|
| 296 |
+
# In[28]:
|
| 297 |
+
|
| 298 |
+
|
| 299 |
+
sc.tl.dendrogram(adata,'leiden',use_rep='scaled|original|X_pca')
|
| 300 |
+
sc.tl.rank_genes_groups(adata, 'leiden', use_rep='scaled|original|X_pca',
|
| 301 |
+
method='t-test',use_raw=False,key_added='leiden_ttest')
|
| 302 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 303 |
+
cmap='Spectral_r',key='leiden_ttest',
|
| 304 |
+
standard_scale='var',n_genes=3)
|
| 305 |
+
|
| 306 |
+
|
| 307 |
+
# cosg is also considered to be a better algorithm for finding marker genes. Here, omicverse provides the calculation of cosg
|
| 308 |
+
#
|
| 309 |
+
# Paper: [Accurate and fast cell marker gene identification with COSG](https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab579/6511197?redirectedFrom=fulltext)
|
| 310 |
+
#
|
| 311 |
+
# Code: https://github.com/genecell/COSG
|
| 312 |
+
#
|
| 313 |
+
|
| 314 |
+
# In[29]:
|
| 315 |
+
|
| 316 |
+
|
| 317 |
+
sc.tl.rank_genes_groups(adata, groupby='leiden',
|
| 318 |
+
method='t-test',use_rep='scaled|original|X_pca',)
|
| 319 |
+
ov.single.cosg(adata, key_added='leiden_cosg', groupby='leiden')
|
| 320 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 321 |
+
cmap='Spectral_r',key='leiden_cosg',
|
| 322 |
+
standard_scale='var',n_genes=3)
|
| 323 |
+
|
| 324 |
+
|
| 325 |
+
# ## Other plotting
|
| 326 |
+
#
|
| 327 |
+
# Next, let's try another chart, which we call the Stacked Volcano Chart. We need to prepare two dictionaries, a `data_dict` and a `color_dict`, both of which have the same key requirements.
|
| 328 |
+
#
|
| 329 |
+
# For `data_dict`. we require the contents within each key to be a DataFrame containing ['names','logfoldchanges','pvals_adj'], where names stands for gene names, logfoldchanges stands for differential expression multiplicity, pvals_adj stands for significance p-value
|
| 330 |
+
#
|
| 331 |
+
|
| 332 |
+
# In[51]:
|
| 333 |
+
|
| 334 |
+
|
| 335 |
+
data_dict={}
|
| 336 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 337 |
+
data_dict[i]=sc.get.rank_genes_groups_df(adata, group=i, key='leiden_ttest',
|
| 338 |
+
pval_cutoff=None,log2fc_min=None)
|
| 339 |
+
|
| 340 |
+
|
| 341 |
+
# In[65]:
|
| 342 |
+
|
| 343 |
+
|
| 344 |
+
data_dict.keys()
|
| 345 |
+
|
| 346 |
+
|
| 347 |
+
# In[64]:
|
| 348 |
+
|
| 349 |
+
|
| 350 |
+
data_dict[i].head()
|
| 351 |
+
|
| 352 |
+
|
| 353 |
+
# For `color_dict`, we require that the colour to be displayed for the current key is stored within each key.`
|
| 354 |
+
|
| 355 |
+
# In[63]:
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
type_color_dict=dict(zip(adata.obs['leiden'].cat.categories,
|
| 359 |
+
adata.uns['leiden_colors']))
|
| 360 |
+
type_color_dict
|
| 361 |
+
|
| 362 |
+
|
| 363 |
+
# There are a number of parameters available here for us to customise the settings. Note that when drawing stacking_vol with omicverse version less than 1.4.13, there is a bug that the vertical coordinate is constant at [-15,15], so we have added some code in this tutorial for visualisation.
|
| 364 |
+
#
|
| 365 |
+
# - data_dict: dict, in each key, there is a dataframe with columns of ['logfoldchanges','pvals_adj','names']
|
| 366 |
+
# - color_dict: dict, in each key, there is a color for each omic
|
| 367 |
+
# - pval_threshold: float, pvalue threshold for significant genes
|
| 368 |
+
# - log2fc_threshold: float, log2fc threshold for significant genes
|
| 369 |
+
# - figsize: tuple, figure size
|
| 370 |
+
# - sig_color: str, color for significant genes
|
| 371 |
+
# - normal_color: str, color for non-significant genes
|
| 372 |
+
# - plot_genes_num: int, number of genes to plot
|
| 373 |
+
# - plot_genes_fontsize: int, fontsize for gene names
|
| 374 |
+
# - plot_genes_weight: str, weight for gene names
|
| 375 |
+
|
| 376 |
+
# In[62]:
|
| 377 |
+
|
| 378 |
+
|
| 379 |
+
fig,axes=ov.utils.stacking_vol(data_dict,type_color_dict,
|
| 380 |
+
pval_threshold=0.01,
|
| 381 |
+
log2fc_threshold=2,
|
| 382 |
+
figsize=(8,4),
|
| 383 |
+
sig_color='#a51616',
|
| 384 |
+
normal_color='#c7c7c7',
|
| 385 |
+
plot_genes_num=2,
|
| 386 |
+
plot_genes_fontsize=6,
|
| 387 |
+
plot_genes_weight='bold',
|
| 388 |
+
)
|
| 389 |
+
|
| 390 |
+
#The following code will be removed in future
|
| 391 |
+
y_min,y_max=0,0
|
| 392 |
+
for i in data_dict.keys():
|
| 393 |
+
y_min=min(y_min,data_dict[i]['logfoldchanges'].min())
|
| 394 |
+
y_max=max(y_max,data_dict[i]['logfoldchanges'].max())
|
| 395 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 396 |
+
axes[i].set_ylim(y_min,y_max)
|
| 397 |
+
plt.suptitle('Stacking_vol',fontsize=12)
|
| 398 |
+
|
| 399 |
+
|
| 400 |
+
# In[ ]:
|
| 401 |
+
|
| 402 |
+
|
| 403 |
+
|
| 404 |
+
|
ovrawm/t_preprocess_gpu.txt
ADDED
|
@@ -0,0 +1,416 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Preprocessing the data of scRNA-seq with omicverse[GPU]
|
| 5 |
+
#
|
| 6 |
+
# The count table, a numeric matrix of genes × cells, is the basic input data structure in the analysis of single-cell RNA-sequencing data. A common preprocessing step is to adjust the counts for variable sampling efficiency and to transform them so that the variance is similar across the dynamic range.
|
| 7 |
+
#
|
| 8 |
+
# Suitable methods to preprocess the scRNA-seq is important. Here, we introduce some preprocessing step to help researchers can perform downstream analysis easyier.
|
| 9 |
+
#
|
| 10 |
+
# User can compare our tutorial with [scanpy'tutorial](https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html) to learn how to use omicverse well
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1DXLSls_ppgJmAaZTUvqazNC_E7EDCxUe?usp=sharing
|
| 13 |
+
|
| 14 |
+
# ## Installation
|
| 15 |
+
#
|
| 16 |
+
# Note that the GPU module is not directly present and needs to be installed separately, for a detailed [tutorial](https://rapids-singlecell.readthedocs.io/en/latest/index.html) see [https://rapids-singlecell.readthedocs.io/en/latest/index.html](https://rapids-singlecell.readthedocs.io/en/latest/index.html)
|
| 17 |
+
#
|
| 18 |
+
# ### pip
|
| 19 |
+
# ```shell
|
| 20 |
+
# pip install rapids-singlecell
|
| 21 |
+
# #rapids
|
| 22 |
+
# pip install \
|
| 23 |
+
# --extra-index-url=https://pypi.nvidia.com \
|
| 24 |
+
# cudf-cu12==24.4.* dask-cudf-cu12==24.4.* cuml-cu12==24.4.* \
|
| 25 |
+
# cugraph-cu12==24.4.* cuspatial-cu12==24.4.* cuproj-cu12==24.4.* \
|
| 26 |
+
# cuxfilter-cu12==24.4.* cucim-cu12==24.4.* pylibraft-cu12==24.4.* \
|
| 27 |
+
# raft-dask-cu12==24.4.* cuvs-cu12==24.4.*
|
| 28 |
+
# #cupy
|
| 29 |
+
# pip install cupy-cuda12x
|
| 30 |
+
# ```
|
| 31 |
+
#
|
| 32 |
+
# ### conda-env
|
| 33 |
+
# Note that in order to avoid conflicts, we will consider installing rapid_singlecell first before installing omicverse.
|
| 34 |
+
#
|
| 35 |
+
# The easiest way to install rapids-singlecell is to use one of the yaml file provided in the [conda](https://github.com/Starlitnightly/omicverse/tree/master/conda) folder. These yaml files install everything needed to run the example notebooks and get you started.
|
| 36 |
+
# ```shell
|
| 37 |
+
# conda env create -f conda/omicverse_gpu.yml
|
| 38 |
+
# # or
|
| 39 |
+
# mamba env create -f conda/omicverse_gpu.yml
|
| 40 |
+
# ```
|
| 41 |
+
|
| 42 |
+
# In[1]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
import omicverse as ov
|
| 46 |
+
import scanpy as sc
|
| 47 |
+
ov.plot_set()
|
| 48 |
+
ov.settings.gpu_init()
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
# The data consist of 3k PBMCs from a Healthy Donor and are freely available from 10x Genomics ([here](http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz) from this [webpage](https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k)). On a unix system, you can uncomment and run the following to download and unpack the data. The last line creates a directory for writing processed data.
|
| 52 |
+
|
| 53 |
+
# In[2]:
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
# !mkdir data
|
| 57 |
+
#!wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 58 |
+
#!cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
|
| 59 |
+
# !mkdir write
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
# In[3]:
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
adata = sc.read_10x_mtx(
|
| 66 |
+
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
|
| 67 |
+
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
|
| 68 |
+
cache=True) # write a cache file for faster subsequent reading
|
| 69 |
+
adata
|
| 70 |
+
|
| 71 |
+
|
| 72 |
+
# In[4]:
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
adata.var_names_make_unique()
|
| 76 |
+
adata.obs_names_make_unique()
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# ## Preprocessing
|
| 80 |
+
#
|
| 81 |
+
# ### Quantity control
|
| 82 |
+
#
|
| 83 |
+
# For single-cell data, we require quality control prior to analysis, including the removal of cells containing double cells, low-expressing cells, and low-expressing genes. In addition to this, we need to filter based on mitochondrial gene ratios, number of transcripts, number of genes expressed per cell, cellular Complexity, etc. For a detailed description of the different QCs please see the document: https://hbctraining.github.io/scRNA-seq/lessons/04_SC_quality_control.html
|
| 84 |
+
|
| 85 |
+
# In[5]:
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
ov.pp.anndata_to_GPU(adata)
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# In[6]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.qc(adata,\n tresh={'mito_perc': 0.2, 'nUMIs': 500, 'detected_genes': 250},\n batch_key=None)\nadata\n")
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# ### High variable Gene Detection
|
| 98 |
+
#
|
| 99 |
+
# Here we try to use Pearson's method to calculate highly variable genes. This is the method that is proposed to be superior to ordinary normalisation. See [Article](https://www.nature.com/articles/s41592-023-01814-1#MOESM3) in *Nature Method* for details.
|
| 100 |
+
#
|
| 101 |
+
|
| 102 |
+
# normalize|HVGs:We use | to control the preprocessing step, | before for the normalisation step, either `shiftlog` or `pearson`, and | after for the highly variable gene calculation step, either `pearson` or `seurat`. Our default is `shiftlog|pearson`.
|
| 103 |
+
#
|
| 104 |
+
# - if you use `mode`=`shiftlog|pearson` you need to set `target_sum=50*1e4`, more people like to se `target_sum=1e4`, we test the result think 50*1e4 will be better
|
| 105 |
+
# - if you use `mode`=`pearson|pearson`, you don't need to set `target_sum`
|
| 106 |
+
#
|
| 107 |
+
# <div class="admonition warning">
|
| 108 |
+
# <p class="admonition-title">Note</p>
|
| 109 |
+
# <p>
|
| 110 |
+
# if the version of `omicverse` lower than `1.4.13`, the mode can only be set between `scanpy` and `pearson`.
|
| 111 |
+
# </p>
|
| 112 |
+
# </div>
|
| 113 |
+
#
|
| 114 |
+
|
| 115 |
+
# In[7]:
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
get_ipython().run_cell_magic('time', '', "adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)\nadata\n")
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# Set the .raw attribute of the AnnData object to the normalized and logarithmized raw gene expression for later use in differential testing and visualizations of gene expression. This simply freezes the state of the AnnData object.
|
| 122 |
+
|
| 123 |
+
# In[8]:
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
adata.raw = adata
|
| 127 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 128 |
+
adata
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
# ## Principal component analysis
|
| 132 |
+
#
|
| 133 |
+
# In contrast to scanpy, we do not directly scale the variance of the original expression matrix, but store the results of the variance scaling in the layer, due to the fact that scale may cause changes in the data distribution, and we have not found scale to be meaningful in any scenario other than a principal component analysis
|
| 134 |
+
|
| 135 |
+
# In[9]:
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
get_ipython().run_cell_magic('time', '', 'ov.pp.scale(adata)\nadata\n')
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
# If you want to perform pca in normlog layer, you can set `layer`=`normlog`, but we think scaled is necessary in PCA.
|
| 142 |
+
|
| 143 |
+
# In[10]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
get_ipython().run_cell_magic('time', '', "ov.pp.pca(adata,layer='scaled',n_pcs=50)\nadata\n")
|
| 147 |
+
|
| 148 |
+
|
| 149 |
+
# In[11]:
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
adata.obsm['X_pca']=adata.obsm['scaled|original|X_pca']
|
| 153 |
+
ov.utils.embedding(adata,
|
| 154 |
+
basis='X_pca',
|
| 155 |
+
color='CST3',
|
| 156 |
+
frameon='small')
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# ## Embedding the neighborhood graph
|
| 160 |
+
#
|
| 161 |
+
# We suggest embedding the graph in two dimensions using UMAP (McInnes et al., 2018), see below. It is potentially more faithful to the global connectivity of the manifold than tSNE, i.e., it better preserves trajectories. In some ocassions, you might still observe disconnected clusters and similar connectivity violations. They can usually be remedied by running:
|
| 162 |
+
|
| 163 |
+
# In[23]:
|
| 164 |
+
|
| 165 |
+
|
| 166 |
+
get_ipython().run_cell_magic('time', '', "ov.pp.neighbors(adata, n_neighbors=15, n_pcs=50,\n use_rep='scaled|original|X_pca',method='cagra')\n")
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
# To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated.
|
| 170 |
+
|
| 171 |
+
# In[19]:
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
adata.obsm["X_mde"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 175 |
+
adata
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
# In[20]:
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
ov.pl.embedding(adata,
|
| 182 |
+
basis='X_mde',
|
| 183 |
+
color='CST3',
|
| 184 |
+
frameon='small')
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# You also can use `umap` to visualize the neighborhood graph
|
| 188 |
+
|
| 189 |
+
# In[21]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
ov.pp.umap(adata)
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
# In[22]:
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
ov.pl.embedding(adata,
|
| 199 |
+
basis='X_umap',
|
| 200 |
+
color='CST3',
|
| 201 |
+
frameon='small')
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
# ## Clustering the neighborhood graph
|
| 205 |
+
#
|
| 206 |
+
# As with Seurat and many other frameworks, we recommend the Leiden graph-clustering method (community detection based on optimizing modularity) by Traag *et al.* (2018). Note that Leiden clustering directly clusters the neighborhood graph of cells, which we already computed in the previous section.
|
| 207 |
+
|
| 208 |
+
# In[24]:
|
| 209 |
+
|
| 210 |
+
|
| 211 |
+
ov.pp.leiden(adata)
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
# In[30]:
|
| 215 |
+
|
| 216 |
+
|
| 217 |
+
ov.pp.anndata_to_CPU(adata)
|
| 218 |
+
|
| 219 |
+
|
| 220 |
+
# We redesigned the visualisation of embedding to distinguish it from scanpy's embedding by adding the parameter `fraemon='small'`, which causes the axes to be scaled with the colourbar
|
| 221 |
+
|
| 222 |
+
# In[25]:
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
ov.pl.embedding(adata,
|
| 226 |
+
basis='X_mde',
|
| 227 |
+
color=['leiden', 'CST3', 'NKG7'],
|
| 228 |
+
frameon='small')
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
# We also provide a boundary visualisation function `ov.utils.plot_ConvexHull` to visualise specific clusters.
|
| 232 |
+
#
|
| 233 |
+
# Arguments:
|
| 234 |
+
# - color: if None will use the color of clusters
|
| 235 |
+
# - alpha: default is 0.2
|
| 236 |
+
|
| 237 |
+
# In[26]:
|
| 238 |
+
|
| 239 |
+
|
| 240 |
+
import matplotlib.pyplot as plt
|
| 241 |
+
fig,ax=plt.subplots( figsize = (4,4))
|
| 242 |
+
|
| 243 |
+
ov.pl.embedding(adata,
|
| 244 |
+
basis='X_mde',
|
| 245 |
+
color=['leiden'],
|
| 246 |
+
frameon='small',
|
| 247 |
+
show=False,
|
| 248 |
+
ax=ax)
|
| 249 |
+
|
| 250 |
+
ov.pl.ConvexHull(adata,
|
| 251 |
+
basis='X_mde',
|
| 252 |
+
cluster_key='leiden',
|
| 253 |
+
hull_cluster='0',
|
| 254 |
+
ax=ax)
|
| 255 |
+
|
| 256 |
+
|
| 257 |
+
# If you have too many labels, e.g. too many cell types, and you are concerned about cell overlap, then consider trying the `ov.utils.gen_mpl_labels` function, which improves text overlap.
|
| 258 |
+
# In addition, we make use of the `patheffects` function, which makes our text have outlines
|
| 259 |
+
#
|
| 260 |
+
# - adjust_kwargs: it could be found in package `adjusttext`
|
| 261 |
+
# - text_kwargs: it could be found in class `plt.texts`
|
| 262 |
+
|
| 263 |
+
# In[27]:
|
| 264 |
+
|
| 265 |
+
|
| 266 |
+
from matplotlib import patheffects
|
| 267 |
+
import matplotlib.pyplot as plt
|
| 268 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 269 |
+
|
| 270 |
+
ov.pl.embedding(adata,
|
| 271 |
+
basis='X_mde',
|
| 272 |
+
color=['leiden'],
|
| 273 |
+
show=False, legend_loc=None, add_outline=False,
|
| 274 |
+
frameon='small',legend_fontoutline=2,ax=ax
|
| 275 |
+
)
|
| 276 |
+
|
| 277 |
+
ov.utils.gen_mpl_labels(
|
| 278 |
+
adata,
|
| 279 |
+
'leiden',
|
| 280 |
+
exclude=("None",),
|
| 281 |
+
basis='X_mde',
|
| 282 |
+
ax=ax,
|
| 283 |
+
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
|
| 284 |
+
text_kwargs=dict(fontsize= 12 ,weight='bold',
|
| 285 |
+
path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),
|
| 286 |
+
)
|
| 287 |
+
|
| 288 |
+
|
| 289 |
+
# In[28]:
|
| 290 |
+
|
| 291 |
+
|
| 292 |
+
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
|
| 293 |
+
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
|
| 294 |
+
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
|
| 295 |
+
|
| 296 |
+
|
| 297 |
+
# In[29]:
|
| 298 |
+
|
| 299 |
+
|
| 300 |
+
sc.pl.dotplot(adata, marker_genes, groupby='leiden',
|
| 301 |
+
standard_scale='var');
|
| 302 |
+
|
| 303 |
+
|
| 304 |
+
# ## Finding marker genes
|
| 305 |
+
#
|
| 306 |
+
# Let us compute a ranking for the highly differential genes in each cluster. For this, by default, the .raw attribute of AnnData is used in case it has been initialized before. The simplest and fastest method to do so is the t-test.
|
| 307 |
+
|
| 308 |
+
# In[31]:
|
| 309 |
+
|
| 310 |
+
|
| 311 |
+
sc.tl.dendrogram(adata,'leiden',use_rep='scaled|original|X_pca')
|
| 312 |
+
sc.tl.rank_genes_groups(adata, 'leiden', use_rep='scaled|original|X_pca',
|
| 313 |
+
method='t-test',use_raw=False,key_added='leiden_ttest')
|
| 314 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 315 |
+
cmap='Spectral_r',key='leiden_ttest',
|
| 316 |
+
standard_scale='var',n_genes=3)
|
| 317 |
+
|
| 318 |
+
|
| 319 |
+
# cosg is also considered to be a better algorithm for finding marker genes. Here, omicverse provides the calculation of cosg
|
| 320 |
+
#
|
| 321 |
+
# Paper: [Accurate and fast cell marker gene identification with COSG](https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab579/6511197?redirectedFrom=fulltext)
|
| 322 |
+
#
|
| 323 |
+
# Code: https://github.com/genecell/COSG
|
| 324 |
+
#
|
| 325 |
+
|
| 326 |
+
# In[32]:
|
| 327 |
+
|
| 328 |
+
|
| 329 |
+
sc.tl.rank_genes_groups(adata, groupby='leiden',
|
| 330 |
+
method='t-test',use_rep='scaled|original|X_pca',)
|
| 331 |
+
ov.single.cosg(adata, key_added='leiden_cosg', groupby='leiden')
|
| 332 |
+
sc.pl.rank_genes_groups_dotplot(adata,groupby='leiden',
|
| 333 |
+
cmap='Spectral_r',key='leiden_cosg',
|
| 334 |
+
standard_scale='var',n_genes=3)
|
| 335 |
+
|
| 336 |
+
|
| 337 |
+
# ## Other plotting
|
| 338 |
+
#
|
| 339 |
+
# Next, let's try another chart, which we call the Stacked Volcano Chart. We need to prepare two dictionaries, a `data_dict` and a `color_dict`, both of which have the same key requirements.
|
| 340 |
+
#
|
| 341 |
+
# For `data_dict`. we require the contents within each key to be a DataFrame containing ['names','logfoldchanges','pvals_adj'], where names stands for gene names, logfoldchanges stands for differential expression multiplicity, pvals_adj stands for significance p-value
|
| 342 |
+
#
|
| 343 |
+
|
| 344 |
+
# In[33]:
|
| 345 |
+
|
| 346 |
+
|
| 347 |
+
data_dict={}
|
| 348 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 349 |
+
data_dict[i]=sc.get.rank_genes_groups_df(adata, group=i, key='leiden_ttest',
|
| 350 |
+
pval_cutoff=None,log2fc_min=None)
|
| 351 |
+
|
| 352 |
+
|
| 353 |
+
# In[34]:
|
| 354 |
+
|
| 355 |
+
|
| 356 |
+
data_dict.keys()
|
| 357 |
+
|
| 358 |
+
|
| 359 |
+
# In[35]:
|
| 360 |
+
|
| 361 |
+
|
| 362 |
+
data_dict[i].head()
|
| 363 |
+
|
| 364 |
+
|
| 365 |
+
# For `color_dict`, we require that the colour to be displayed for the current key is stored within each key.`
|
| 366 |
+
|
| 367 |
+
# In[36]:
|
| 368 |
+
|
| 369 |
+
|
| 370 |
+
type_color_dict=dict(zip(adata.obs['leiden'].cat.categories,
|
| 371 |
+
adata.uns['leiden_colors']))
|
| 372 |
+
type_color_dict
|
| 373 |
+
|
| 374 |
+
|
| 375 |
+
# There are a number of parameters available here for us to customise the settings. Note that when drawing stacking_vol with omicverse version less than 1.4.13, there is a bug that the vertical coordinate is constant at [-15,15], so we have added some code in this tutorial for visualisation.
|
| 376 |
+
#
|
| 377 |
+
# - data_dict: dict, in each key, there is a dataframe with columns of ['logfoldchanges','pvals_adj','names']
|
| 378 |
+
# - color_dict: dict, in each key, there is a color for each omic
|
| 379 |
+
# - pval_threshold: float, pvalue threshold for significant genes
|
| 380 |
+
# - log2fc_threshold: float, log2fc threshold for significant genes
|
| 381 |
+
# - figsize: tuple, figure size
|
| 382 |
+
# - sig_color: str, color for significant genes
|
| 383 |
+
# - normal_color: str, color for non-significant genes
|
| 384 |
+
# - plot_genes_num: int, number of genes to plot
|
| 385 |
+
# - plot_genes_fontsize: int, fontsize for gene names
|
| 386 |
+
# - plot_genes_weight: str, weight for gene names
|
| 387 |
+
|
| 388 |
+
# In[37]:
|
| 389 |
+
|
| 390 |
+
|
| 391 |
+
fig,axes=ov.utils.stacking_vol(data_dict,type_color_dict,
|
| 392 |
+
pval_threshold=0.01,
|
| 393 |
+
log2fc_threshold=2,
|
| 394 |
+
figsize=(8,4),
|
| 395 |
+
sig_color='#a51616',
|
| 396 |
+
normal_color='#c7c7c7',
|
| 397 |
+
plot_genes_num=2,
|
| 398 |
+
plot_genes_fontsize=6,
|
| 399 |
+
plot_genes_weight='bold',
|
| 400 |
+
)
|
| 401 |
+
|
| 402 |
+
#The following code will be removed in future
|
| 403 |
+
y_min,y_max=0,0
|
| 404 |
+
for i in data_dict.keys():
|
| 405 |
+
y_min=min(y_min,data_dict[i]['logfoldchanges'].min())
|
| 406 |
+
y_max=max(y_max,data_dict[i]['logfoldchanges'].max())
|
| 407 |
+
for i in adata.obs['leiden'].cat.categories:
|
| 408 |
+
axes[i].set_ylim(y_min,y_max)
|
| 409 |
+
plt.suptitle('Stacking_vol',fontsize=12)
|
| 410 |
+
|
| 411 |
+
|
| 412 |
+
# In[ ]:
|
| 413 |
+
|
| 414 |
+
|
| 415 |
+
|
| 416 |
+
|
ovrawm/t_scdeg.txt
ADDED
|
@@ -0,0 +1,316 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Differential expression analysis in single cell
|
| 5 |
+
#
|
| 6 |
+
# Sometimes we need to compare differentially expressed genes or differentially expressed features between two cell types on single cell data, but existing methods focus more on cell-specific gene analysis. Researchers need to transfer bulk RNA-seq analysis to single-cell analysis, which involves interaction between different programming languages or programming tools, adding significantly to the workload of the researcher.
|
| 7 |
+
#
|
| 8 |
+
# Here, we use omicverse's bulk RNA-seq pyDEG method to complete differential expression analysis at the single cell level. We will present two different perspectives, one from the perspective of all cells and one from the perspective of the metacellular.
|
| 9 |
+
#
|
| 10 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/12faBRh0xT7v6KSy8NCSRqbegF_AEoDXr?usp=sharing
|
| 11 |
+
|
| 12 |
+
# In[1]:
|
| 13 |
+
|
| 14 |
+
|
| 15 |
+
import omicverse as ov
|
| 16 |
+
import scanpy as sc
|
| 17 |
+
import scvelo as scv
|
| 18 |
+
|
| 19 |
+
ov.utils.ov_plot_set()
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
# ## Data preprocessed
|
| 23 |
+
#
|
| 24 |
+
# We need to normalized and scale the data at first.
|
| 25 |
+
|
| 26 |
+
# In[2]:
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
adata = scv.datasets.pancreas()
|
| 30 |
+
adata
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
# In[3]:
|
| 34 |
+
|
| 35 |
+
|
| 36 |
+
adata.X.max()
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
# We found that the max value of anndata object larger than 10 and type is int. We need to normalize and log1p it
|
| 40 |
+
|
| 41 |
+
# In[4]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
#quantity control
|
| 45 |
+
adata=ov.pp.qc(adata,
|
| 46 |
+
tresh={'mito_perc': 0.05, 'nUMIs': 500, 'detected_genes': 250})
|
| 47 |
+
#normalize and high variable genes (HVGs) calculated
|
| 48 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 49 |
+
|
| 50 |
+
#save the whole genes and filter the non-HVGs
|
| 51 |
+
adata.raw = adata
|
| 52 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 53 |
+
|
| 54 |
+
#scale the adata.X
|
| 55 |
+
ov.pp.scale(adata)
|
| 56 |
+
|
| 57 |
+
#Dimensionality Reduction
|
| 58 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 59 |
+
|
| 60 |
+
|
| 61 |
+
# In[5]:
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
adata.X.max()
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# ## Different expression in total level
|
| 68 |
+
#
|
| 69 |
+
# We then select the target cells to be analysed, including `Alpha` and `Beta`, derive the expression matrix using `to_df()` and build the differential expression analysis module using `pyDEG`
|
| 70 |
+
|
| 71 |
+
# In[6]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
test_adata=adata[adata.obs['clusters'].isin(['Alpha','Beta'])]
|
| 75 |
+
test_adata
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# In[7]:
|
| 79 |
+
|
| 80 |
+
|
| 81 |
+
dds=ov.bulk.pyDEG(test_adata.to_df(layer='lognorm').T)
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
# In[8]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
dds.drop_duplicates_index()
|
| 88 |
+
print('... drop_duplicates_index success')
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# We also need to set up an experimental group and a control group, i.e. the two types of cells to be compared and analysed
|
| 92 |
+
|
| 93 |
+
# In[9]:
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
treatment_groups=test_adata.obs[test_adata.obs['clusters']=='Alpha'].index.tolist()
|
| 97 |
+
control_groups=test_adata.obs[test_adata.obs['clusters']=='Beta'].index.tolist()
|
| 98 |
+
result=dds.deg_analysis(treatment_groups,control_groups,method='ttest')
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
# In[10]:
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
result.sort_values('qvalue').head()
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
# In[11]:
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
# -1 means automatically calculates
|
| 111 |
+
dds.foldchange_set(fc_threshold=-1,
|
| 112 |
+
pval_threshold=0.05,
|
| 113 |
+
logp_max=10)
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
# In[12]:
|
| 117 |
+
|
| 118 |
+
|
| 119 |
+
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
|
| 120 |
+
plot_genes_num=8,plot_genes_fontsize=12,)
|
| 121 |
+
|
| 122 |
+
|
| 123 |
+
# In[13]:
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
dds.plot_boxplot(genes=['Irx1','Adra2a'],treatment_groups=treatment_groups,
|
| 127 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 128 |
+
legend_bbox=(2,0.55))
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
# In[14]:
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
ov.utils.embedding(adata,
|
| 135 |
+
basis='X_umap',
|
| 136 |
+
frameon='small',
|
| 137 |
+
color=['clusters','Irx1','Adra2a'])
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
# ## Different expression in Metacells level
|
| 141 |
+
#
|
| 142 |
+
# Here, we calculated the metacells from the whole scRNA-seq datasets using SEACells, and the same analyze with total level.
|
| 143 |
+
|
| 144 |
+
# ### Constructing a metacellular object
|
| 145 |
+
#
|
| 146 |
+
# We can use `ov.single.MetaCell` to construct a metacellular object to train the SEACells model, the arguments can be found in below.
|
| 147 |
+
#
|
| 148 |
+
# - :param ad: (AnnData) annotated data matrix
|
| 149 |
+
# - :param build_kernel_on: (str) key corresponding to matrix in ad.obsm which is used to compute kernel for metacells
|
| 150 |
+
# Typically 'X_pca' for scRNA or 'X_svd' for scATAC
|
| 151 |
+
# - :param n_SEACells: (int) number of SEACells to compute
|
| 152 |
+
# - :param use_gpu: (bool) whether to use GPU for computation
|
| 153 |
+
# - :param verbose: (bool) whether to suppress verbose program logging
|
| 154 |
+
# - :param n_waypoint_eigs: (int) number of eigenvectors to use for waypoint initialization
|
| 155 |
+
# - :param n_neighbors: (int) number of nearest neighbors to use for graph construction
|
| 156 |
+
# - :param convergence_epsilon: (float) convergence threshold for Franke-Wolfe algorithm
|
| 157 |
+
# - :param l2_penalty: (float) L2 penalty for Franke-Wolfe algorithm
|
| 158 |
+
# - :param max_franke_wolfe_iters: (int) maximum number of iterations for Franke-Wolfe algorithm
|
| 159 |
+
# - :param use_sparse: (bool) whether to use sparse matrix operations. Currently only supported for CPU implementation.
|
| 160 |
+
|
| 161 |
+
# In[15]:
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
meta_obj=ov.single.MetaCell(adata,use_rep='scaled|original|X_pca',n_metacells=150,
|
| 165 |
+
use_gpu=True)
|
| 166 |
+
|
| 167 |
+
|
| 168 |
+
# In[16]:
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
meta_obj.initialize_archetypes()
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# ## Train and save the model
|
| 175 |
+
|
| 176 |
+
# In[17]:
|
| 177 |
+
|
| 178 |
+
|
| 179 |
+
meta_obj.train(min_iter=10, max_iter=50)
|
| 180 |
+
|
| 181 |
+
|
| 182 |
+
# In[34]:
|
| 183 |
+
|
| 184 |
+
|
| 185 |
+
meta_obj.save('seacells/model.pkl')
|
| 186 |
+
|
| 187 |
+
|
| 188 |
+
# In[ ]:
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
meta_obj.load('seacells/model.pkl')
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
# ## Predicted the metacells
|
| 195 |
+
#
|
| 196 |
+
# we can use `predicted` to predicted the metacells of raw scRNA-seq data. There are two method can be selected, one is `soft`, the other is `hard`.
|
| 197 |
+
#
|
| 198 |
+
# In the `soft` method, Aggregates cells within each SEACell, summing over all raw data x assignment weight for all cells belonging to a SEACell. Data is un-normalized and pseudo-raw aggregated counts are stored in .layers['raw']. Attributes associated with variables (.var) are copied over, but relevant per SEACell attributes must be manually copied, since certain attributes may need to be summed, or averaged etc, depending on the attribute.
|
| 199 |
+
#
|
| 200 |
+
# In the `hard` method, Aggregates cells within each SEACell, summing over all raw data for all cells belonging to a SEACell. Data is unnormalized and raw aggregated counts are stored .layers['raw']. Attributes associated with variables (.var) are copied over, but relevant per SEACell attributes must be manually copied, since certain attributes may need to be summed, or averaged etc, depending on the attribute.
|
| 201 |
+
|
| 202 |
+
# In[19]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
ad=meta_obj.predicted(method='soft',celltype_label='clusters',
|
| 206 |
+
summarize_layer='lognorm')
|
| 207 |
+
|
| 208 |
+
|
| 209 |
+
# In[20]:
|
| 210 |
+
|
| 211 |
+
|
| 212 |
+
ad.X.min(),ad.X.max()
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
# In[21]:
|
| 216 |
+
|
| 217 |
+
|
| 218 |
+
import matplotlib.pyplot as plt
|
| 219 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 220 |
+
ov.utils.embedding(
|
| 221 |
+
meta_obj.adata,
|
| 222 |
+
basis="X_umap",
|
| 223 |
+
color=['clusters'],
|
| 224 |
+
frameon='small',
|
| 225 |
+
title="Meta cells",
|
| 226 |
+
#legend_loc='on data',
|
| 227 |
+
legend_fontsize=14,
|
| 228 |
+
legend_fontoutline=2,
|
| 229 |
+
size=10,
|
| 230 |
+
ax=ax,
|
| 231 |
+
alpha=0.2,
|
| 232 |
+
#legend_loc='',
|
| 233 |
+
add_outline=False,
|
| 234 |
+
#add_outline=True,
|
| 235 |
+
outline_color='black',
|
| 236 |
+
outline_width=1,
|
| 237 |
+
show=False,
|
| 238 |
+
#palette=ov.utils.blue_color[:],
|
| 239 |
+
#legend_fontweight='normal'
|
| 240 |
+
)
|
| 241 |
+
ov.single._metacell.plot_metacells(ax,meta_obj.adata,color='#CB3E35',
|
| 242 |
+
)
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
# ### Differentially expressed analysis
|
| 246 |
+
#
|
| 247 |
+
# Similar to total cells for differential expression analysis, we used metacells for differential expression in the same way.
|
| 248 |
+
|
| 249 |
+
# In[23]:
|
| 250 |
+
|
| 251 |
+
|
| 252 |
+
test_adata=ad[ad.obs['celltype'].isin(['Alpha','Beta'])]
|
| 253 |
+
test_adata
|
| 254 |
+
|
| 255 |
+
|
| 256 |
+
# In[24]:
|
| 257 |
+
|
| 258 |
+
|
| 259 |
+
dds_meta=ov.bulk.pyDEG(test_adata.to_df().T)
|
| 260 |
+
|
| 261 |
+
|
| 262 |
+
# In[25]:
|
| 263 |
+
|
| 264 |
+
|
| 265 |
+
dds_meta.drop_duplicates_index()
|
| 266 |
+
print('... drop_duplicates_index success')
|
| 267 |
+
|
| 268 |
+
|
| 269 |
+
# We also need to set up an experimental group and a control group, i.e. the two types of cells to be compared and analysed
|
| 270 |
+
|
| 271 |
+
# In[27]:
|
| 272 |
+
|
| 273 |
+
|
| 274 |
+
treatment_groups=test_adata.obs[test_adata.obs['celltype']=='Alpha'].index.tolist()
|
| 275 |
+
control_groups=test_adata.obs[test_adata.obs['celltype']=='Beta'].index.tolist()
|
| 276 |
+
result=dds_meta.deg_analysis(treatment_groups,control_groups,method='ttest')
|
| 277 |
+
|
| 278 |
+
|
| 279 |
+
# In[28]:
|
| 280 |
+
|
| 281 |
+
|
| 282 |
+
result.sort_values('qvalue').head()
|
| 283 |
+
|
| 284 |
+
|
| 285 |
+
# In[29]:
|
| 286 |
+
|
| 287 |
+
|
| 288 |
+
# -1 means automatically calculates
|
| 289 |
+
dds_meta.foldchange_set(fc_threshold=-1,
|
| 290 |
+
pval_threshold=0.05,
|
| 291 |
+
logp_max=10)
|
| 292 |
+
|
| 293 |
+
|
| 294 |
+
# In[30]:
|
| 295 |
+
|
| 296 |
+
|
| 297 |
+
dds_meta.plot_volcano(title='DEG Analysis',figsize=(4,4),
|
| 298 |
+
plot_genes_num=8,plot_genes_fontsize=12,)
|
| 299 |
+
|
| 300 |
+
|
| 301 |
+
# In[31]:
|
| 302 |
+
|
| 303 |
+
|
| 304 |
+
dds_meta.plot_boxplot(genes=['Ctxn2','Mnx1'],treatment_groups=treatment_groups,
|
| 305 |
+
control_groups=control_groups,figsize=(2,3),fontsize=12,
|
| 306 |
+
legend_bbox=(2,0.55))
|
| 307 |
+
|
| 308 |
+
|
| 309 |
+
# In[32]:
|
| 310 |
+
|
| 311 |
+
|
| 312 |
+
ov.utils.embedding(adata,
|
| 313 |
+
basis='X_umap',
|
| 314 |
+
frameon='small',
|
| 315 |
+
color=['clusters','Ctxn2','Mnx1'])
|
| 316 |
+
|
ovrawm/t_scdrug.txt
ADDED
|
@@ -0,0 +1,225 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Drug response predict with scDrug
|
| 5 |
+
#
|
| 6 |
+
# scDrug is a database that can be used to predict the drug sensitivity of single cells based on an existing database of drug responses. In the downstream tasks of single cell analysis, especially in tumours, we are fully interested in potential drugs and combination therapies. To this end, we have integrated scDrug's IC50 prediction and inferCNV to infer the function of tumour cells to build a drug screening pipeline.
|
| 7 |
+
#
|
| 8 |
+
# Paper: [scDrug: From single-cell RNA-seq to drug response prediction](https://www.sciencedirect.com/science/article/pii/S2001037022005505)
|
| 9 |
+
#
|
| 10 |
+
# Code: https://github.com/ailabstw/scDrug
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1mayoMO7I7qjYIRjrZEi8r5zuERcxAEcF?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
import scanpy as sc
|
| 19 |
+
import infercnvpy as cnv
|
| 20 |
+
import matplotlib.pyplot as plt
|
| 21 |
+
import os
|
| 22 |
+
|
| 23 |
+
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
|
| 24 |
+
sc.settings.set_figure_params(dpi=80, facecolor='white')
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# ## Infer the Tumor from scRNA-seq
|
| 28 |
+
#
|
| 29 |
+
# Here we use Infercnvpy's example data to complete the tumour analysis, you can also refer to the official tutorial for this step: https://infercnvpy.readthedocs.io/en/latest/notebooks/tutorial_3k.html
|
| 30 |
+
#
|
| 31 |
+
# So, we provide a utility function ov.utils.get_gene_annotation to supplement the coordinate information from GTF files. The following usage assumes that the adata.var_names correspond to “gene_name” attribute in the GTF file. For other cases, please check the function documentation.
|
| 32 |
+
#
|
| 33 |
+
# The GTF file used here can be downloaded from [GENCODE](http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M25/).
|
| 34 |
+
#
|
| 35 |
+
# T2T-CHM13 gtf file can be download from [figshare](https://figshare.com/ndownloader/files/40628072)
|
| 36 |
+
|
| 37 |
+
# In[3]:
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
adata = cnv.datasets.maynard2020_3k()
|
| 41 |
+
|
| 42 |
+
ov.utils.get_gene_annotation(
|
| 43 |
+
adata, gtf="gencode.v43.basic.annotation.gtf.gz",
|
| 44 |
+
gtf_by="gene_name"
|
| 45 |
+
)
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# In[ ]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
adata=adata[:,~adata.var['chrom'].isnull()]
|
| 52 |
+
adata.var['chromosome']=adata.var['chrom']
|
| 53 |
+
adata.var['start']=adata.var['chromStart']
|
| 54 |
+
adata.var['end']=adata.var['chromEnd']
|
| 55 |
+
adata.var['ensg']=adata.var['gene_id']
|
| 56 |
+
adata.var.loc[:, ["ensg", "chromosome", "start", "end"]].head()
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
# We noted that infercnvpy need to normalize and log the matrix at first
|
| 60 |
+
|
| 61 |
+
# In[4]:
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
adata
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# We use the immune cells as reference and infer the cnv score of each cells in scRNA-seq
|
| 68 |
+
|
| 69 |
+
# In[5]:
|
| 70 |
+
|
| 71 |
+
|
| 72 |
+
# We provide all immune cell types as "normal cells".
|
| 73 |
+
cnv.tl.infercnv(
|
| 74 |
+
adata,
|
| 75 |
+
reference_key="cell_type",
|
| 76 |
+
reference_cat=[
|
| 77 |
+
"B cell",
|
| 78 |
+
"Macrophage",
|
| 79 |
+
"Mast cell",
|
| 80 |
+
"Monocyte",
|
| 81 |
+
"NK cell",
|
| 82 |
+
"Plasma cell",
|
| 83 |
+
"T cell CD4",
|
| 84 |
+
"T cell CD8",
|
| 85 |
+
"T cell regulatory",
|
| 86 |
+
"mDC",
|
| 87 |
+
"pDC",
|
| 88 |
+
],
|
| 89 |
+
window_size=250,
|
| 90 |
+
)
|
| 91 |
+
cnv.tl.pca(adata)
|
| 92 |
+
cnv.pp.neighbors(adata)
|
| 93 |
+
cnv.tl.leiden(adata)
|
| 94 |
+
cnv.tl.umap(adata)
|
| 95 |
+
cnv.tl.cnv_score(adata)
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# In[6]:
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
sc.pl.umap(adata, color="cnv_score", show=False)
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
# We set an appropriate threshold for the cnv_score, here we set it to 0.03 and identify cells greater than 0.03 as tumour cells
|
| 105 |
+
|
| 106 |
+
# In[7]:
|
| 107 |
+
|
| 108 |
+
|
| 109 |
+
adata.obs["cnv_status"] = "normal"
|
| 110 |
+
adata.obs.loc[
|
| 111 |
+
adata.obs["cnv_score"]>0.03, "cnv_status"
|
| 112 |
+
] = "tumor"
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# In[8]:
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
sc.pl.umap(adata, color="cnv_status", show=False)
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
# We extracted tumour cells separately for drug prediction response
|
| 122 |
+
|
| 123 |
+
# In[11]:
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
tumor=adata[adata.obs['cnv_status']=='tumor']
|
| 127 |
+
tumor.X.max()
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# ## Tumor preprocessing
|
| 131 |
+
#
|
| 132 |
+
# We need to extract the highly variable genes in the tumour for further analysis, and found out the sub-cluster in tumor
|
| 133 |
+
|
| 134 |
+
# In[12]:
|
| 135 |
+
|
| 136 |
+
|
| 137 |
+
adata=tumor
|
| 138 |
+
print('Preprocessing...')
|
| 139 |
+
sc.pp.filter_cells(adata, min_genes=200)
|
| 140 |
+
sc.pp.filter_genes(adata, min_cells=3)
|
| 141 |
+
adata.var['mt'] = adata.var_names.str.startswith('MT-')
|
| 142 |
+
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
|
| 143 |
+
if not (adata.obs.pct_counts_mt == 0).all():
|
| 144 |
+
adata = adata[adata.obs.pct_counts_mt < 30, :]
|
| 145 |
+
|
| 146 |
+
adata.raw = adata.copy()
|
| 147 |
+
|
| 148 |
+
sc.pp.highly_variable_genes(adata)
|
| 149 |
+
adata = adata[:, adata.var.highly_variable]
|
| 150 |
+
sc.pp.scale(adata)
|
| 151 |
+
sc.tl.pca(adata, svd_solver='arpack')
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
# In[13]:
|
| 155 |
+
|
| 156 |
+
|
| 157 |
+
sc.pp.neighbors(adata, n_pcs=20)
|
| 158 |
+
sc.tl.umap(adata)
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
# Here, we need to download the scDrug database and mods so that the subsequent predictions can be made properly
|
| 162 |
+
|
| 163 |
+
# In[27]:
|
| 164 |
+
|
| 165 |
+
|
| 166 |
+
ov.utils.download_GDSC_data()
|
| 167 |
+
ov.utils.download_CaDRReS_model()
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
# Then, we apply Single-Cell Data Analysis once again to carry out sub-clustering on the tumor clusters at automatically determined resolution.
|
| 171 |
+
|
| 172 |
+
# In[18]:
|
| 173 |
+
|
| 174 |
+
|
| 175 |
+
adata, res,plot_df = ov.single.autoResolution(adata,cpus=4)
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
# Don't forget to save your data
|
| 179 |
+
|
| 180 |
+
# In[20]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
results_file = os.path.join('./', 'scanpyobj.h5ad')
|
| 184 |
+
adata.write(results_file)
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# In[21]:
|
| 188 |
+
|
| 189 |
+
|
| 190 |
+
results_file = os.path.join('./', 'scanpyobj.h5ad')
|
| 191 |
+
adata=sc.read(results_file)
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
# ## IC50 predicted
|
| 195 |
+
#
|
| 196 |
+
# Drug Response Prediction examined scanpyobj.h5ad generated in Single-Cell Data Analysis, reported clusterwise IC50 and cell death percentages to drugs in the GDSC database via CaDRReS-Sc (a recommender system framework for in silico drug response prediction), or drug sensitivity AUC in the PRISM database from [DepMap Portal PRISM-19Q4](https://doi.org/10.1038/s43018-019-0018-6).
|
| 197 |
+
#
|
| 198 |
+
# Note we need to download the CaDRReS-Sc from github by `git clone https://github.com/CSB5/CaDRReS-Sc`
|
| 199 |
+
|
| 200 |
+
# In[24]:
|
| 201 |
+
|
| 202 |
+
|
| 203 |
+
get_ipython().system('git clone https://github.com/CSB5/CaDRReS-Sc')
|
| 204 |
+
|
| 205 |
+
|
| 206 |
+
# To run drug response predicted, we need to set:
|
| 207 |
+
#
|
| 208 |
+
# - scriptpath: the CaDRReS-Sc path we downloaded just now
|
| 209 |
+
# - modelpath: the model path we downloaded just now
|
| 210 |
+
# - output: the save path of drug response predicted result
|
| 211 |
+
|
| 212 |
+
# In[25]:
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
import ov
|
| 216 |
+
job=ov.single.Drug_Response(adata,scriptpath='CaDRReS-Sc',
|
| 217 |
+
modelpath='models/',
|
| 218 |
+
output='result')
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
# In[ ]:
|
| 222 |
+
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
|
ovrawm/t_scmulan.txt
ADDED
|
@@ -0,0 +1,199 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# ## Using scMulan to annotate cell types in Heart, Lung, Liver, Bone marrow, Blood, Brain, and Thymus
|
| 5 |
+
|
| 6 |
+
# In this study, the authors enrich the pre-training paradigm by integrating an abundance of metadata and a multiplicity of pre-training tasks, and obtain scMulan, a multitask generative pre-trained language model tailored for single-cell analysis. They represent a cell as a structured cell sentence (c-sentence) by encoding its gene expression, metadata terms, and target tasks as words of tuples, each consisting of entities and their corresponding values. They construct a unified generative framework to model the cell language on c-sentence and design three pretraining tasks to bridge the microscopic and macroscopic information within the c-sentences. They pre-train scMulan on 10 million single-cell transcriptomic data and their corresponding metadata, with 368 million parameters. As a single model, scMulan can accomplish tasks zero-shot for cell type annotation, batch integration, and conditional cell generation, guided by different task prompts.
|
| 7 |
+
|
| 8 |
+
# #### we provide a liver dataset sampled (percentage of 20%) from Suo C, 2022 (doi/10.1126/science.abo0510)
|
| 9 |
+
# **Paper:** [scMulan: a multitask generative pre-trained language model for single-cell analysis](https://www.biorxiv.org/content/10.1101/2024.01.25.577152v1)
|
| 10 |
+
# **Data download:** https://cloud.tsinghua.edu.cn/f/45a7fd2a27e543539f59/?dl=1
|
| 11 |
+
# **Pre-train model download:** https://cloud.tsinghua.edu.cn/f/2250c5df51034b2e9a85/?dl=1
|
| 12 |
+
#
|
| 13 |
+
# If you found this tutorial helpful, please cite scMulan and OmicVerse:
|
| 14 |
+
# Bian H, Chen Y, Dong X, et al. scMulan: a multitask generative pre-trained language model for single-cell analysis[C]//International Conference on Research in Computational Molecular Biology. Cham: Springer Nature Switzerland, 2024: 479-482.
|
| 15 |
+
|
| 16 |
+
# In[36]:
|
| 17 |
+
|
| 18 |
+
|
| 19 |
+
import os
|
| 20 |
+
#os.environ["CUDA_VISIBLE_DEVICES"] = "-1" # if use CPU only
|
| 21 |
+
import scanpy as sc
|
| 22 |
+
import omicverse as ov
|
| 23 |
+
ov.plot_set()
|
| 24 |
+
#import scMulan
|
| 25 |
+
#from scMulan import GeneSymbolUniform
|
| 26 |
+
|
| 27 |
+
|
| 28 |
+
# ## 1. load h5ad
|
| 29 |
+
# You can download the liver dataset from the following link: https://cloud.tsinghua.edu.cn/f/45a7fd2a27e543539f59/?dl=1
|
| 30 |
+
#
|
| 31 |
+
# It's recommended that you use h5ad here with raw count (and after your QC)
|
| 32 |
+
#
|
| 33 |
+
|
| 34 |
+
# In[4]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
adata = sc.read('./data/liver_test.h5ad')
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
# In[5]:
|
| 41 |
+
|
| 42 |
+
|
| 43 |
+
adata
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
# In[6]:
|
| 47 |
+
|
| 48 |
+
|
| 49 |
+
from scipy.sparse import csc_matrix
|
| 50 |
+
adata.X = csc_matrix(adata.X)
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
# ## 2. transform original h5ad with uniformed genes (42117 genes)
|
| 54 |
+
|
| 55 |
+
# This step transform the genes in input adata to 42117 gene symbols and reserves the corresponding gene expression values. The gene symbols are the same as the pre-trained scMulan model.
|
| 56 |
+
|
| 57 |
+
# In[7]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
adata_GS_uniformed = ov.externel.scMulan.GeneSymbolUniform(input_adata=adata,
|
| 61 |
+
output_dir="./data",
|
| 62 |
+
output_prefix='liver')
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
# ## 3. process uniformed data (simply norm and log1p)
|
| 66 |
+
|
| 67 |
+
# In[8]:
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
## you can read the saved uniformed adata
|
| 71 |
+
|
| 72 |
+
adata_GS_uniformed=sc.read_h5ad('./data/liver_uniformed.h5ad')
|
| 73 |
+
|
| 74 |
+
|
| 75 |
+
# In[9]:
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
adata_GS_uniformed
|
| 79 |
+
|
| 80 |
+
|
| 81 |
+
# In[10]:
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
# norm and log1p count matrix
|
| 85 |
+
# in some case, the count matrix is not normalized, and log1p is not applied.
|
| 86 |
+
# So we need to normalize the count matrix
|
| 87 |
+
if adata_GS_uniformed.X.max() > 10:
|
| 88 |
+
sc.pp.normalize_total(adata_GS_uniformed, target_sum=1e4)
|
| 89 |
+
sc.pp.log1p(adata_GS_uniformed)
|
| 90 |
+
|
| 91 |
+
|
| 92 |
+
# ## 4. load scMulan
|
| 93 |
+
|
| 94 |
+
# In[11]:
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# you should first download ckpt from https://cloud.tsinghua.edu.cn/f/2250c5df51034b2e9a85/?dl=1
|
| 98 |
+
# put it under .ckpt/ckpt_scMulan.pt
|
| 99 |
+
# by: wget https://cloud.tsinghua.edu.cn/f/2250c5df51034b2e9a85/?dl=1 -O ckpt/ckpt_scMulan.pt
|
| 100 |
+
|
| 101 |
+
ckp_path = './ckpt/ckpt_scMulan.pt'
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
# In[12]:
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
scml = ov.externel.scMulan.model_inference(ckp_path, adata_GS_uniformed)
|
| 108 |
+
base_process = scml.cuda_count()
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
# In[13]:
|
| 112 |
+
|
| 113 |
+
|
| 114 |
+
scml.get_cell_types_and_embds_for_adata(parallel=True, n_process = 1)
|
| 115 |
+
# scml.get_cell_types_and_embds_for_adata(parallel=False) # for only using CPU, but it is really slow.
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# The predicted cell types are stored in scml.adata.obs['cell_type_from_scMulan'], besides the cell embeddings (for multibatch integration) in scml.adata.obsm['X_scMulan'] (not used in this tutorial).
|
| 119 |
+
|
| 120 |
+
# ## 5. visualization
|
| 121 |
+
#
|
| 122 |
+
# Here, we visualize the cell types predicted by scMulan. And we also visualize the original cell types in the dataset.
|
| 123 |
+
|
| 124 |
+
# In[14]:
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
adata_mulan = scml.adata.copy()
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# In[15]:
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
# calculated the 2-D embedding of the adata using pyMDE
|
| 134 |
+
ov.pp.scale(adata_mulan)
|
| 135 |
+
ov.pp.pca(adata_mulan)
|
| 136 |
+
|
| 137 |
+
#sc.pl.pca_variance_ratio(adata_mulan)
|
| 138 |
+
ov.pp.mde(adata_mulan,embedding_dim=2,n_neighbors=15, basis='X_mde',
|
| 139 |
+
n_pcs=10, use_rep='scaled|original|X_pca',)
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
# In[26]:
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
# Here, we can see the cell type annotation from scMulan
|
| 146 |
+
ov.pl.embedding(adata_mulan,basis='X_mde',
|
| 147 |
+
color=["cell_type_from_scMulan",],
|
| 148 |
+
ncols=1,frameon='small')
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
# In[29]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
adata_mulan.obsm['X_umap']=adata_mulan.obsm['X_mde']
|
| 155 |
+
|
| 156 |
+
|
| 157 |
+
# In[30]:
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
# you can run smoothing function to filter the false positives
|
| 161 |
+
ov.externel.scMulan.cell_type_smoothing(adata_mulan, threshold=0.1)
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
# In[31]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# cell_type_from_mulan_smoothing: pred+smoothing
|
| 168 |
+
# cell_type: original annotations by the authors
|
| 169 |
+
ov.pl.embedding(adata_mulan,basis='X_mde',
|
| 170 |
+
color=["cell_type_from_mulan_smoothing","cell_type"],
|
| 171 |
+
ncols=1,frameon='small')
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# In[32]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
adata_mulan
|
| 178 |
+
|
| 179 |
+
|
| 180 |
+
# In[33]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
top_celltypes = adata_mulan.obs.cell_type_from_scMulan.value_counts().index[:20]
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
# In[34]:
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# you can select some cell types of interest (from scMulan's prediction) for visulization
|
| 190 |
+
# selected_cell_types = ["NK cell", "Kupffer cell", "Conventional dendritic cell 2"] # as example
|
| 191 |
+
selected_cell_types = top_celltypes
|
| 192 |
+
ov.externel.scMulan.visualize_selected_cell_types(adata_mulan,selected_cell_types,smoothing=True)
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
# In[ ]:
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
|
| 199 |
+
|
ovrawm/t_simba.txt
ADDED
|
@@ -0,0 +1,146 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Data integration and batch correction with SIMBA
|
| 5 |
+
#
|
| 6 |
+
# Here we will use three scRNA-seq human pancreas datasets of different studies as an example to illustrate how SIMBA performs scRNA-seq batch correction for multiple batches
|
| 7 |
+
#
|
| 8 |
+
# We follow the corresponding tutorial at [SIMBA](https://simba-bio.readthedocs.io/en/latest/rna_human_pancreas.html). We do not provide much explanation, and instead refer to the original tutorial.
|
| 9 |
+
#
|
| 10 |
+
# Paper: [SIMBA: single-cell embedding along with features](https://www.nature.com/articles/s41592-023-01899-8)
|
| 11 |
+
#
|
| 12 |
+
# Code: https://github.com/huidongchen/simba
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
from omicverse.utils import mde
|
| 19 |
+
workdir = 'result_human_pancreas'
|
| 20 |
+
ov.utils.ov_plot_set()
|
| 21 |
+
|
| 22 |
+
|
| 23 |
+
# We need to install simba at first
|
| 24 |
+
#
|
| 25 |
+
# ```
|
| 26 |
+
# conda install -c bioconda simba
|
| 27 |
+
# ```
|
| 28 |
+
#
|
| 29 |
+
# or
|
| 30 |
+
#
|
| 31 |
+
# ```
|
| 32 |
+
# pip install git+https://github.com/huidongchen/simba
|
| 33 |
+
# pip install git+https://github.com/pinellolab/simba_pbg
|
| 34 |
+
# ```
|
| 35 |
+
|
| 36 |
+
# ## Read data
|
| 37 |
+
#
|
| 38 |
+
# The anndata object was concat from three anndata in simba: `simba.datasets.rna_baron2016()`, `simba.datasets.rna_segerstolpe2016()`, and `simba.datasets.rna_muraro2016()`
|
| 39 |
+
#
|
| 40 |
+
# It can be downloaded from figshare: https://figshare.com/ndownloader/files/41418600
|
| 41 |
+
|
| 42 |
+
# In[2]:
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
adata=ov.utils.read('simba_adata_raw.h5ad')
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# We need to set workdir to initiate the pySIMBA object
|
| 49 |
+
|
| 50 |
+
# In[3]:
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
simba_object=ov.single.pySIMBA(adata,workdir)
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
# ## Preprocess
|
| 57 |
+
#
|
| 58 |
+
# Follow the raw tutorial, we set the paragument as default.
|
| 59 |
+
|
| 60 |
+
# In[4]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
simba_object.preprocess(batch_key='batch',min_n_cells=3,
|
| 64 |
+
method='lib_size',n_top_genes=3000,n_bins=5)
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# ## Generate a graph for training
|
| 68 |
+
#
|
| 69 |
+
# Observations and variables within each Anndata object are both represented as nodes (entities).
|
| 70 |
+
#
|
| 71 |
+
# the data store in `simba_object.uns['simba_batch_edge_dict']`
|
| 72 |
+
|
| 73 |
+
# In[5]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
simba_object.gen_graph()
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# ## PBG training
|
| 80 |
+
#
|
| 81 |
+
# Before training, let’s take a look at the current parameters:
|
| 82 |
+
#
|
| 83 |
+
# - dict_config['workers'] = 12 #The number of CPUs.
|
| 84 |
+
|
| 85 |
+
# In[10]:
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
simba_object.train(num_workers=6)
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# In[6]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
simba_object.load('result_human_pancreas/pbg/graph0')
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# ## Batch correction
|
| 98 |
+
#
|
| 99 |
+
# Here, we use `simba_object.batch_correction()` to perform the batch correction
|
| 100 |
+
#
|
| 101 |
+
# <div class="admonition note">
|
| 102 |
+
# <p class="admonition-title">Note</p>
|
| 103 |
+
# <p>
|
| 104 |
+
# If the batch is greater than 10, then the batch correction is less effective
|
| 105 |
+
# </p>
|
| 106 |
+
# </div>
|
| 107 |
+
|
| 108 |
+
# In[7]:
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
adata=simba_object.batch_correction()
|
| 112 |
+
adata
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# ## Visualize
|
| 116 |
+
#
|
| 117 |
+
# We also use `mde` instead `umap` to visualize the result
|
| 118 |
+
|
| 119 |
+
# In[8]:
|
| 120 |
+
|
| 121 |
+
|
| 122 |
+
adata.obsm["X_mde"] = mde(adata.obsm["X_simba"])
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
# In[11]:
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
sc.pl.embedding(adata,basis='X_mde',color=['cell_type1','batch'])
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
# Certainly, umap can also be used to visualize
|
| 132 |
+
|
| 133 |
+
# In[10]:
|
| 134 |
+
|
| 135 |
+
|
| 136 |
+
import scanpy as sc
|
| 137 |
+
sc.pp.neighbors(adata, use_rep="X_simba")
|
| 138 |
+
sc.tl.umap(adata)
|
| 139 |
+
sc.pl.umap(adata,color=['cell_type1','batch'])
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
# In[ ]:
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
|
ovrawm/t_single_batch.txt
ADDED
|
@@ -0,0 +1,333 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Data integration and batch correction
|
| 5 |
+
#
|
| 6 |
+
# An important task of single-cell analysis is the integration of several samples, which we can perform with omicverse.
|
| 7 |
+
#
|
| 8 |
+
# Here we demonstrate how to merge data using omicverse and perform a corrective analysis for batch effects. We provide a total of 4 methods for batch effect correction in omicverse, including harmony, scanorama and combat which do not require GPU, and SIMBA which requires GPU. if available, we recommend using GPU-based scVI and scANVI to get the best batch effect correction results.
|
| 9 |
+
#
|
| 10 |
+
#
|
| 11 |
+
|
| 12 |
+
# In[1]:
|
| 13 |
+
|
| 14 |
+
|
| 15 |
+
import omicverse as ov
|
| 16 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 17 |
+
import scanpy as sc
|
| 18 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 19 |
+
ov.utils.ov_plot_set()
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
# ## Data integration
|
| 23 |
+
#
|
| 24 |
+
# First, we need to concat the data of scRNA-seq from different batch. We can use `sc.concat` to perform it。
|
| 25 |
+
#
|
| 26 |
+
# The dataset we will use to demonstrate data integration contains several samples of bone marrow mononuclear cells. These samples were originally created for the Open Problems in Single-Cell Analysis NeurIPS Competition 2021.
|
| 27 |
+
#
|
| 28 |
+
# We selected sample of `s1d3`, `s2d1` and `s3d7` to perform integrate. The individual data can be downloaded from figshare.
|
| 29 |
+
#
|
| 30 |
+
# - s1d3:
|
| 31 |
+
# - s2d1:
|
| 32 |
+
# - s3d7:
|
| 33 |
+
|
| 34 |
+
# In[2]:
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
adata1=ov.read('neurips2021_s1d3.h5ad')
|
| 38 |
+
adata1.obs['batch']='s1d3'
|
| 39 |
+
adata2=ov.read('neurips2021_s2d1.h5ad')
|
| 40 |
+
adata2.obs['batch']='s2d1'
|
| 41 |
+
adata3=ov.read('neurips2021_s3d7.h5ad')
|
| 42 |
+
adata3.obs['batch']='s3d7'
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# In[3]:
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
adata=sc.concat([adata1,adata2,adata3],merge='same')
|
| 49 |
+
adata
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
# We can see that there are now three elements in the batch
|
| 53 |
+
|
| 54 |
+
# In[4]:
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
adata.obs['batch'].unique()
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
# In[7]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
import numpy as np
|
| 64 |
+
adata.X=adata.X.astype(np.int64)
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# ## Data preprocess and Batch visualize
|
| 68 |
+
#
|
| 69 |
+
# We first performed quality control of the data and normalisation with screening for highly variable genes. Then visualise potential batch effects in the data.
|
| 70 |
+
#
|
| 71 |
+
# Here, we can set `batch_key=batch` to correct the doublet detectation and Highly variable genes identifcation.
|
| 72 |
+
|
| 73 |
+
# In[8]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
adata=ov.pp.qc(adata,
|
| 77 |
+
tresh={'mito_perc': 0.2, 'nUMIs': 500, 'detected_genes': 250},
|
| 78 |
+
batch_key='batch')
|
| 79 |
+
adata
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
# We can store the raw counts if we need the raw counts after filtered the HVGs.
|
| 83 |
+
|
| 84 |
+
# In[10]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',
|
| 88 |
+
n_HVGs=3000,batch_key=None)
|
| 89 |
+
adata
|
| 90 |
+
|
| 91 |
+
|
| 92 |
+
# In[11]:
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
adata.raw = adata
|
| 96 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 97 |
+
adata
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# We can save the pre-processed data.
|
| 101 |
+
|
| 102 |
+
# In[12]:
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
adata.write_h5ad('neurips2021_batch_normlog.h5ad',compression='gzip')
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
# Similarly, we calculated PCA for HVGs and visualised potential batch effects in the data using pymde. pymde is GPU-accelerated UMAP.
|
| 109 |
+
|
| 110 |
+
# In[13]:
|
| 111 |
+
|
| 112 |
+
|
| 113 |
+
ov.pp.scale(adata)
|
| 114 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50,mask_var='highly_variable_features')
|
| 115 |
+
|
| 116 |
+
adata.obsm["X_mde_pca"] = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
|
| 117 |
+
|
| 118 |
+
|
| 119 |
+
# There is a very clear batch effect in the data
|
| 120 |
+
|
| 121 |
+
# In[14]:
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
ov.utils.embedding(adata,
|
| 125 |
+
basis='X_mde_pca',frameon='small',
|
| 126 |
+
color=['batch','cell_type'],show=False)
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
# ## Harmony
|
| 130 |
+
#
|
| 131 |
+
# Harmony is an algorithm for performing integration of single cell genomics datasets. Please check out manuscript on [Nature Methods](https://www.nature.com/articles/s41592-019-0619-0).
|
| 132 |
+
#
|
| 133 |
+
# 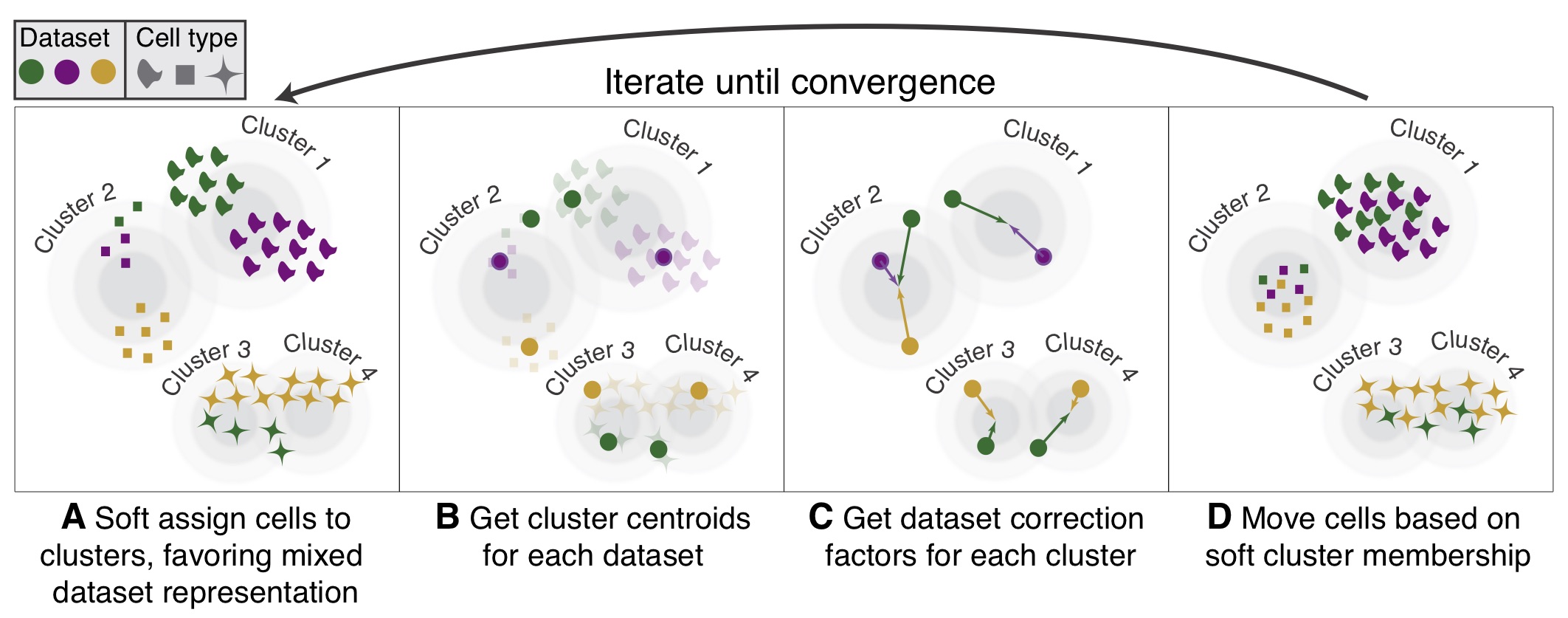
|
| 134 |
+
|
| 135 |
+
# The function `ov.single.batch_correction` can be set in three methods: `harmony`,`combat` and `scanorama`
|
| 136 |
+
|
| 137 |
+
# In[40]:
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
adata_harmony=ov.single.batch_correction(adata,batch_key='batch',
|
| 141 |
+
methods='harmony',n_pcs=50)
|
| 142 |
+
adata
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
# In[41]:
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
adata.obsm["X_mde_harmony"] = ov.utils.mde(adata.obsm["X_harmony"])
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
# In[42]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
ov.utils.embedding(adata,
|
| 155 |
+
basis='X_mde_harmony',frameon='small',
|
| 156 |
+
color=['batch','cell_type'],show=False)
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# ## Combat
|
| 160 |
+
#
|
| 161 |
+
# combat is a batch effect correction method that is very widely used in bulk RNA-seq, and it works just as well on single-cell sequencing data.
|
| 162 |
+
#
|
| 163 |
+
#
|
| 164 |
+
|
| 165 |
+
# In[43]:
|
| 166 |
+
|
| 167 |
+
|
| 168 |
+
adata_combat=ov.single.batch_correction(adata,batch_key='batch',
|
| 169 |
+
methods='combat',n_pcs=50)
|
| 170 |
+
adata
|
| 171 |
+
|
| 172 |
+
|
| 173 |
+
# In[44]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
adata.obsm["X_mde_combat"] = ov.utils.mde(adata.obsm["X_combat"])
|
| 177 |
+
|
| 178 |
+
|
| 179 |
+
# In[45]:
|
| 180 |
+
|
| 181 |
+
|
| 182 |
+
ov.utils.embedding(adata,
|
| 183 |
+
basis='X_mde_combat',frameon='small',
|
| 184 |
+
color=['batch','cell_type'],show=False)
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# ## scanorama
|
| 188 |
+
#
|
| 189 |
+
# Integration of single-cell RNA sequencing (scRNA-seq) data from multiple experiments, laboratories and technologies can uncover biological insights, but current methods for scRNA-seq data integration are limited by a requirement for datasets to derive from functionally similar cells. We present Scanorama, an algorithm that identifies and merges the shared cell types among all pairs of datasets and accurately integrates heterogeneous collections of scRNA-seq data.
|
| 190 |
+
#
|
| 191 |
+
# 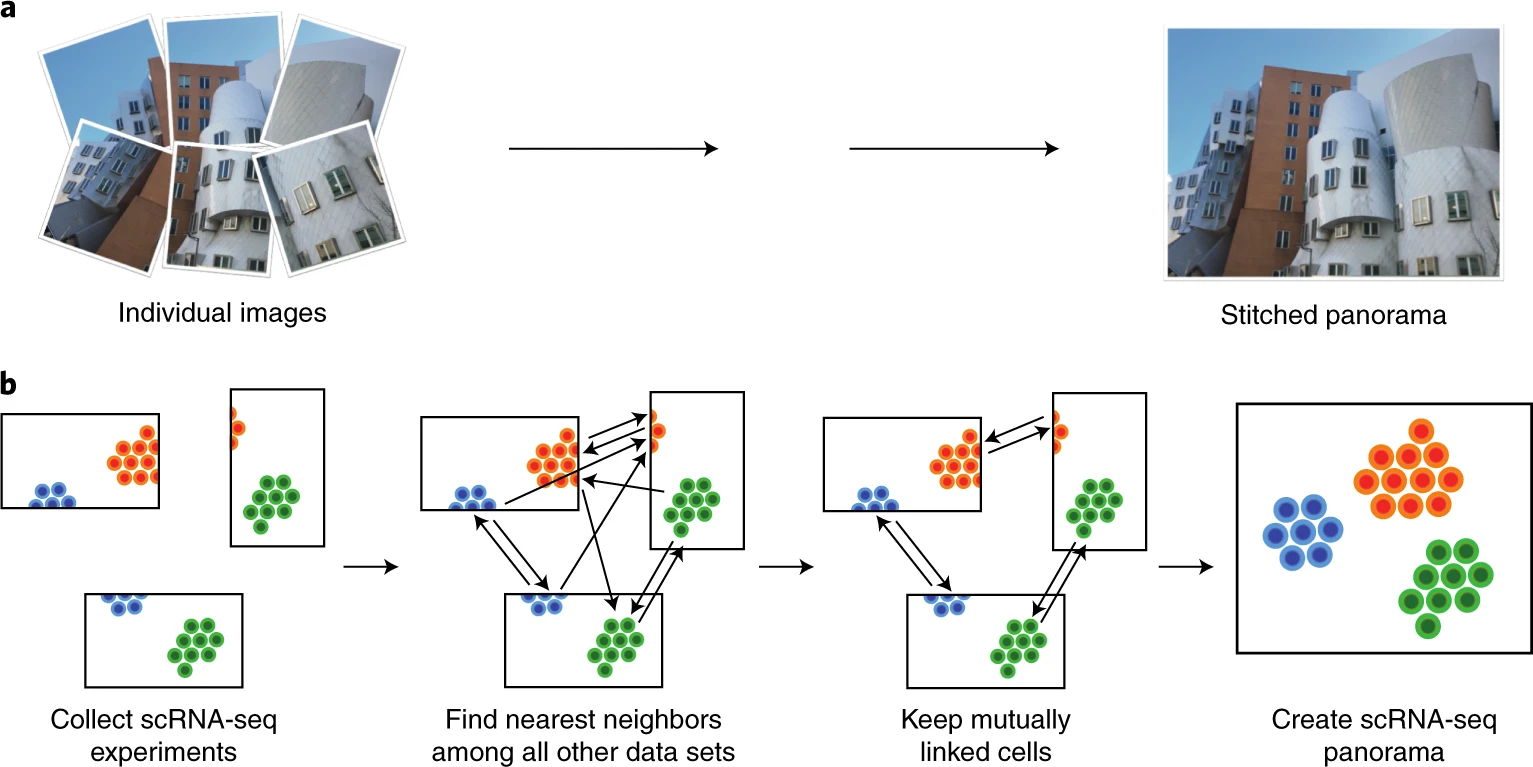
|
| 192 |
+
|
| 193 |
+
# In[46]:
|
| 194 |
+
|
| 195 |
+
|
| 196 |
+
adata_scanorama=ov.single.batch_correction(adata,batch_key='batch',
|
| 197 |
+
methods='scanorama',n_pcs=50)
|
| 198 |
+
adata
|
| 199 |
+
|
| 200 |
+
|
| 201 |
+
# In[47]:
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
adata.obsm["X_mde_scanorama"] = ov.utils.mde(adata.obsm["X_scanorama"])
|
| 205 |
+
|
| 206 |
+
|
| 207 |
+
# In[48]:
|
| 208 |
+
|
| 209 |
+
|
| 210 |
+
ov.utils.embedding(adata,
|
| 211 |
+
basis='X_mde_scanorama',frameon='small',
|
| 212 |
+
color=['batch','cell_type'],show=False)
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
# ## scVI
|
| 216 |
+
#
|
| 217 |
+
# An important task of single-cell analysis is the integration of several samples, which we can perform with scVI. For integration, scVI treats the data as unlabelled. When our dataset is fully labelled (perhaps in independent studies, or independent analysis pipelines), we can obtain an integration that better preserves biology using scANVI, which incorporates cell type annotation information. Here we demonstrate this functionality with an integrated analysis of cells from the lung atlas integration task from the scIB manuscript. The same pipeline would generally be used to analyze any collection of scRNA-seq datasets.
|
| 218 |
+
|
| 219 |
+
# In[3]:
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
adata_scvi=ov.single.batch_correction(adata,batch_key='batch',
|
| 223 |
+
methods='scVI',n_layers=2, n_latent=30, gene_likelihood="nb")
|
| 224 |
+
adata
|
| 225 |
+
|
| 226 |
+
|
| 227 |
+
# In[4]:
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
adata.obsm["X_mde_scVI"] = ov.utils.mde(adata.obsm["X_scVI"])
|
| 231 |
+
|
| 232 |
+
|
| 233 |
+
# In[5]:
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
ov.utils.embedding(adata,
|
| 237 |
+
basis='X_mde_scVI',frameon='small',
|
| 238 |
+
color=['batch','cell_type'],show=False)
|
| 239 |
+
|
| 240 |
+
|
| 241 |
+
# ## MIRA+CODAL
|
| 242 |
+
#
|
| 243 |
+
# Topic modeling of batched single-cell data is challenging because these models cannot typically distinguish between biological and technical effects of the assay. CODAL (COvariate Disentangling Augmented Loss) uses a novel mutual information regularization technique to explicitly disentangle these two sources of variation.
|
| 244 |
+
|
| 245 |
+
# In[15]:
|
| 246 |
+
|
| 247 |
+
|
| 248 |
+
LDA_obj=ov.utils.LDA_topic(adata,feature_type='expression',
|
| 249 |
+
highly_variable_key='highly_variable_features',
|
| 250 |
+
layers='counts',batch_key='batch',learning_rate=1e-3)
|
| 251 |
+
|
| 252 |
+
|
| 253 |
+
# In[16]:
|
| 254 |
+
|
| 255 |
+
|
| 256 |
+
LDA_obj.plot_topic_contributions(6)
|
| 257 |
+
|
| 258 |
+
|
| 259 |
+
# In[17]:
|
| 260 |
+
|
| 261 |
+
|
| 262 |
+
LDA_obj.predicted(15)
|
| 263 |
+
|
| 264 |
+
|
| 265 |
+
# In[37]:
|
| 266 |
+
|
| 267 |
+
|
| 268 |
+
adata.obsm["X_mde_mira_topic"] = ov.utils.mde(adata.obsm["X_topic_compositions"])
|
| 269 |
+
adata.obsm["X_mde_mira_feature"] = ov.utils.mde(adata.obsm["X_umap_features"])
|
| 270 |
+
|
| 271 |
+
|
| 272 |
+
# In[38]:
|
| 273 |
+
|
| 274 |
+
|
| 275 |
+
ov.utils.embedding(adata,
|
| 276 |
+
basis='X_mde_mira_topic',frameon='small',
|
| 277 |
+
color=['batch','cell_type'],show=False)
|
| 278 |
+
|
| 279 |
+
|
| 280 |
+
# In[39]:
|
| 281 |
+
|
| 282 |
+
|
| 283 |
+
ov.utils.embedding(adata,
|
| 284 |
+
basis='X_mde_mira_feature',frameon='small',
|
| 285 |
+
color=['batch','cell_type'],show=False)
|
| 286 |
+
|
| 287 |
+
|
| 288 |
+
# ## Benchmarking test
|
| 289 |
+
#
|
| 290 |
+
# The methods demonstrated here are selected based on results from benchmarking experiments including the single-cell integration benchmarking project [Luecken et al., 2021]. This project also produced a software package called [scib](https://www.github.com/theislab/scib) that can be used to run a range of integration methods as well as the metrics that were used for evaluation. In this section, we show how to use this package to evaluate the quality of an integration.
|
| 291 |
+
|
| 292 |
+
# In[6]:
|
| 293 |
+
|
| 294 |
+
|
| 295 |
+
adata.write_h5ad('neurips2021_batch_all.h5ad',compression='gzip')
|
| 296 |
+
|
| 297 |
+
|
| 298 |
+
# In[2]:
|
| 299 |
+
|
| 300 |
+
|
| 301 |
+
adata=sc.read('neurips2021_batch_all.h5ad')
|
| 302 |
+
|
| 303 |
+
|
| 304 |
+
# In[7]:
|
| 305 |
+
|
| 306 |
+
|
| 307 |
+
adata.obsm['X_pca']=adata.obsm['scaled|original|X_pca'].copy()
|
| 308 |
+
adata.obsm['X_mira_topic']=adata.obsm['X_topic_compositions'].copy()
|
| 309 |
+
adata.obsm['X_mira_feature']=adata.obsm['X_umap_features'].copy()
|
| 310 |
+
|
| 311 |
+
|
| 312 |
+
# In[ ]:
|
| 313 |
+
|
| 314 |
+
|
| 315 |
+
from scib_metrics.benchmark import Benchmarker
|
| 316 |
+
bm = Benchmarker(
|
| 317 |
+
adata,
|
| 318 |
+
batch_key="batch",
|
| 319 |
+
label_key="cell_type",
|
| 320 |
+
embedding_obsm_keys=["X_pca", "X_combat", "X_harmony",
|
| 321 |
+
'X_scanorama','X_mira_topic','X_mira_feature','X_scVI'],
|
| 322 |
+
n_jobs=8,
|
| 323 |
+
)
|
| 324 |
+
bm.benchmark()
|
| 325 |
+
|
| 326 |
+
|
| 327 |
+
# In[9]:
|
| 328 |
+
|
| 329 |
+
|
| 330 |
+
bm.plot_results_table(min_max_scale=False)
|
| 331 |
+
|
| 332 |
+
|
| 333 |
+
# We can find that harmony removes the batch effect the best of the three methods that do not use the GPU, scVI is method to remove batch effect using GPU.
|
ovrawm/t_slat.txt
ADDED
|
@@ -0,0 +1,365 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Single cell spatial alignment tools
|
| 5 |
+
#
|
| 6 |
+
# SLAT (Spatially-Linked Alignment Tool), a graph-based algorithm for efficient and effective alignment of spatial slices. Adopting a graph adversarial matching strategy, SLAT is the first algorithm capable of aligning heterogenous spatial data across distinct technologies and modalities.
|
| 7 |
+
#
|
| 8 |
+
# We made two improvements in integrating the STT algorithm in OmicVerse:
|
| 9 |
+
#
|
| 10 |
+
# - **Fix the running error in alignment**: We fixed some issues with the scSLAT package on pypi.
|
| 11 |
+
# - **Added more downstream analysis**: We have expanded on the original tutorial by combining the tutorial and reproduce code given by the authors for downstream analysis.
|
| 12 |
+
#
|
| 13 |
+
# If you found this tutorial helpful, please cite SLAT and OmicVerse:
|
| 14 |
+
#
|
| 15 |
+
# - Xia, CR., Cao, ZJ., Tu, XM. et al. Spatial-linked alignment tool (SLAT) for aligning heterogenous slices. Nat Commun 14, 7236 (2023). https://doi.org/10.1038/s41467-023-43105-5
|
| 16 |
+
|
| 17 |
+
# In[1]:
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
import omicverse as ov
|
| 21 |
+
import os
|
| 22 |
+
|
| 23 |
+
import scanpy as sc
|
| 24 |
+
import numpy as np
|
| 25 |
+
import pandas as pd
|
| 26 |
+
import torch
|
| 27 |
+
ov.plot_set()
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
# In[2]:
|
| 31 |
+
|
| 32 |
+
|
| 33 |
+
#import scSLAT
|
| 34 |
+
from omicverse.externel.scSLAT.model import load_anndatas, Cal_Spatial_Net, run_SLAT, scanpy_workflow, spatial_match
|
| 35 |
+
from omicverse.externel.scSLAT.viz import match_3D_multi, hist, Sankey, match_3D_celltype, Sankey,Sankey_multi,build_3D
|
| 36 |
+
from omicverse.externel.scSLAT.metrics import region_statistics
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
# ## Preprocess Data
|
| 40 |
+
#
|
| 41 |
+
# adata1.h5ad: E11.5 mouse embryo dataset, download from [here](https://drive.google.com/uc?export=download&id=1KkuJt6aSlKS1AJzFZjE_odypY-GINRuD)
|
| 42 |
+
#
|
| 43 |
+
# adata2.h5ad: E12.5 mouse embryo dataset, download from [here](https://drive.google.com/uc?export=download&id=1YIiEmjGfHxcDbGn4nv2kzmTHUo3_q5hJ)
|
| 44 |
+
|
| 45 |
+
# In[3]:
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
adata1 = sc.read_h5ad('data/E115_Stereo.h5ad')
|
| 49 |
+
adata2 = sc.read_h5ad('data/E125_Stereo.h5ad')
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
# In[4]:
|
| 53 |
+
|
| 54 |
+
|
| 55 |
+
adata1.obs['week']='E11.5'
|
| 56 |
+
adata2.obs['week']='E12.5'
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
# In[5]:
|
| 60 |
+
|
| 61 |
+
|
| 62 |
+
sc.pl.spatial(adata1, color='annotation', spot_size=3)
|
| 63 |
+
sc.pl.spatial(adata2, color='annotation', spot_size=3)
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
# ## Run SLAT
|
| 67 |
+
#
|
| 68 |
+
# Then we run SLAT as usual
|
| 69 |
+
|
| 70 |
+
# In[6]:
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
Cal_Spatial_Net(adata1, k_cutoff=20, model='KNN')
|
| 74 |
+
Cal_Spatial_Net(adata2, k_cutoff=20, model='KNN')
|
| 75 |
+
edges, features = load_anndatas([adata1, adata2], feature='DPCA', check_order=False)
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# In[7]:
|
| 79 |
+
|
| 80 |
+
|
| 81 |
+
embd0, embd1, time = run_SLAT(features, edges, LGCN_layer=5)
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
# In[8]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
best, index, distance = spatial_match([embd0, embd1], reorder=False, adatas=[adata1,adata2])
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
# In[9]:
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
matching = np.array([range(index.shape[0]), best])
|
| 94 |
+
best_match = distance[:,0]
|
| 95 |
+
region_statistics(best_match, start=0.5, number_of_interval=10)
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# ## Visualization of alignment
|
| 99 |
+
|
| 100 |
+
# In[10]:
|
| 101 |
+
|
| 102 |
+
|
| 103 |
+
import matplotlib.pyplot as plt
|
| 104 |
+
matching_list=[matching]
|
| 105 |
+
model = build_3D([adata1,adata2], matching_list,subsample_size=300, )
|
| 106 |
+
ax=model.draw_3D(hide_axis=True, line_color='#c2c2c2', height=1, size=[6,6], line_width=1)
|
| 107 |
+
|
| 108 |
+
|
| 109 |
+
# Then we check the alignment quality of the whole slide
|
| 110 |
+
|
| 111 |
+
# In[11]:
|
| 112 |
+
|
| 113 |
+
|
| 114 |
+
adata2.obs['low_quality_index']= best_match
|
| 115 |
+
adata2.obs['low_quality_index'] = adata2.obs['low_quality_index'].astype(float)
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# In[12]:
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
adata2.obsm['spatial']
|
| 122 |
+
|
| 123 |
+
|
| 124 |
+
# In[13]:
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
sc.pl.spatial(adata2, color='low_quality_index', spot_size=3, title='Quality')
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# We use a Sankey diagram to show the correspondence between cell types at different stages of development
|
| 131 |
+
|
| 132 |
+
# In[33]:
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
fig=Sankey_multi(adata_li=[adata1,adata2],
|
| 136 |
+
prefix_li=['E11.5','E12.5'],
|
| 137 |
+
matching_li=[matching],
|
| 138 |
+
clusters='annotation',filter_num=10,
|
| 139 |
+
node_opacity = 0.8,
|
| 140 |
+
link_opacity = 0.2,
|
| 141 |
+
layout=[800,500],
|
| 142 |
+
font_size=12,
|
| 143 |
+
font_color='Black',
|
| 144 |
+
save_name=None,
|
| 145 |
+
format='png',
|
| 146 |
+
width=1200,
|
| 147 |
+
height=1000,
|
| 148 |
+
return_fig=True)
|
| 149 |
+
fig.show()
|
| 150 |
+
|
| 151 |
+
|
| 152 |
+
# In[34]:
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
fig.write_html("slat_sankey.html")
|
| 156 |
+
|
| 157 |
+
|
| 158 |
+
# ## Focus on developing Kidney
|
| 159 |
+
#
|
| 160 |
+
# We highlighted the “Kidney” cells in E12.5 and their aligned precursor cells in E11.5 in alignment results. Consistent with our biological priors, the precursors of the kidney are the mesonephros and the metanephros
|
| 161 |
+
#
|
| 162 |
+
# Then we focus on another organ: ‘Ovary’, and found ovary only has single spatial origin. It is interesting that precursors of ovary are spatially close to the mesonephros (see Kidney part), because mammalian ovary originates from the regressed mesonephros.
|
| 163 |
+
|
| 164 |
+
# In[27]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
color_dict1=dict(zip(adata1.obs['annotation'].cat.categories,
|
| 168 |
+
adata1.uns['annotation_colors'].tolist()))
|
| 169 |
+
adata1_df = pd.DataFrame({'index':range(embd0.shape[0]),
|
| 170 |
+
'x': adata1.obsm['spatial'][:,0],
|
| 171 |
+
'y': adata1.obsm['spatial'][:,1],
|
| 172 |
+
'celltype':adata1.obs['annotation'],
|
| 173 |
+
'color':adata1.obs['annotation'].map(color_dict1)
|
| 174 |
+
}
|
| 175 |
+
)
|
| 176 |
+
color_dict2=dict(zip(adata2.obs['annotation'].cat.categories,
|
| 177 |
+
adata2.uns['annotation_colors'].tolist()))
|
| 178 |
+
adata2_df = pd.DataFrame({'index':range(embd1.shape[0]),
|
| 179 |
+
'x': adata2.obsm['spatial'][:,0],
|
| 180 |
+
'y': adata2.obsm['spatial'][:,1],
|
| 181 |
+
'celltype':adata2.obs['annotation'],
|
| 182 |
+
'color':adata2.obs['annotation'].map(color_dict2)
|
| 183 |
+
}
|
| 184 |
+
)
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# In[28]:
|
| 188 |
+
|
| 189 |
+
|
| 190 |
+
kidney_align = match_3D_celltype(adata1_df, adata2_df, matching, meta='celltype',
|
| 191 |
+
highlight_celltype = [['Urogenital ridge'],['Kidney','Ovary']],
|
| 192 |
+
subsample_size=10000, highlight_line = ['blue'], scale_coordinate = True )
|
| 193 |
+
kidney_align.draw_3D(size= [6, 6], line_width =0.8, point_size=[0.6,0.6], hide_axis=True)
|
| 194 |
+
|
| 195 |
+
|
| 196 |
+
# We can get the lineage of the query's cells and mappings using the following function
|
| 197 |
+
|
| 198 |
+
# In[15]:
|
| 199 |
+
|
| 200 |
+
|
| 201 |
+
def cal_matching_cell(target_adata,query_adata,matching,query_cell,clusters='annotation',):
|
| 202 |
+
adata1_df = pd.DataFrame({'index':range(target_adata.shape[0]),
|
| 203 |
+
'x': target_adata.obsm['spatial'][:,0],
|
| 204 |
+
'y': target_adata.obsm['spatial'][:,1],
|
| 205 |
+
'celltype':target_adata.obs[clusters]})
|
| 206 |
+
adata2_df = pd.DataFrame({'index':range(query_adata.shape[0]),
|
| 207 |
+
'x': query_adata.obsm['spatial'][:,0],
|
| 208 |
+
'y': query_adata.obsm['spatial'][:,1],
|
| 209 |
+
'celltype':query_adata.obs[clusters]})
|
| 210 |
+
query_adata = target_adata[matching[1,adata2_df.loc[adata2_df.celltype==query_cell,'index'].values],:]
|
| 211 |
+
#adata2_df['target_celltype'] = adata1_df.iloc[matching[1,:],:]['celltype'].to_list()
|
| 212 |
+
#adata2_df['target_obs_names'] = adata1_df.iloc[matching[1,:],:].index.to_list()
|
| 213 |
+
|
| 214 |
+
#query_obs=adata2_df.loc[adata2_df['celltype']==query_cell,'target_obs_names'].tolist()
|
| 215 |
+
return query_adata
|
| 216 |
+
|
| 217 |
+
|
| 218 |
+
|
| 219 |
+
# We find that maps mapped on 3D also show up well on 2D
|
| 220 |
+
|
| 221 |
+
# In[21]:
|
| 222 |
+
|
| 223 |
+
|
| 224 |
+
query_adata=cal_matching_cell(target_adata=adata1,
|
| 225 |
+
query_adata=adata2,
|
| 226 |
+
matching=matching,
|
| 227 |
+
query_cell='Kidney',clusters='annotation')
|
| 228 |
+
query_adata
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
# In[17]:
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
adata1.obs['kidney_anno']=''
|
| 235 |
+
adata1.obs.loc[query_adata.obs.index,'kidney_anno']=query_adata.obs['annotation']
|
| 236 |
+
|
| 237 |
+
|
| 238 |
+
# In[18]:
|
| 239 |
+
|
| 240 |
+
|
| 241 |
+
sc.pl.spatial(adata1, color='kidney_anno', spot_size=3,
|
| 242 |
+
palette=['#F5F5F5','#ff7f0e', 'green',])
|
| 243 |
+
|
| 244 |
+
|
| 245 |
+
# We are concerned with Kidney lineage development, so we integrated the cells corresponding to the Kidney lineage on the two sections of E11 and E12, and then we could use the method of difference analysis to study the dynamic process of Kidney lineage development.
|
| 246 |
+
|
| 247 |
+
# In[22]:
|
| 248 |
+
|
| 249 |
+
|
| 250 |
+
kidney_lineage_ad=sc.concat([query_adata,adata2[adata2.obs['annotation']=='Kidney']],merge='same')
|
| 251 |
+
kidney_lineage_ad=ov.pp.preprocess(kidney_lineage_ad,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 252 |
+
kidney_lineage_ad.raw = kidney_lineage_ad
|
| 253 |
+
kidney_lineage_ad = kidney_lineage_ad[:, kidney_lineage_ad.var.highly_variable_features]
|
| 254 |
+
ov.pp.scale(kidney_lineage_ad)
|
| 255 |
+
ov.pp.pca(kidney_lineage_ad)
|
| 256 |
+
ov.pp.neighbors(kidney_lineage_ad,use_rep='scaled|original|X_pca',metric="cosine")
|
| 257 |
+
ov.utils.cluster(kidney_lineage_ad,method='leiden',resolution=1)
|
| 258 |
+
ov.pp.umap(kidney_lineage_ad)
|
| 259 |
+
|
| 260 |
+
|
| 261 |
+
# In[23]:
|
| 262 |
+
|
| 263 |
+
|
| 264 |
+
ov.pl.embedding(kidney_lineage_ad,basis='X_umap',
|
| 265 |
+
color=['annotation','week','leiden'],
|
| 266 |
+
frameon='small')
|
| 267 |
+
|
| 268 |
+
|
| 269 |
+
# In[25]:
|
| 270 |
+
|
| 271 |
+
|
| 272 |
+
# Nphs1 https://www.nature.com/articles/s41467-021-22266-1
|
| 273 |
+
sc.pl.dotplot(kidney_lineage_ad,{'nephron progenitors':['Wnt9b','Osr1','Nphs1','Lhx1','Pax2','Pax8'],
|
| 274 |
+
'metanephric':['Eya1','Shisa3','Foxc1'],
|
| 275 |
+
'kidney':['Wt1','Wnt4','Nr2f2','Dach1','Cd44']} ,
|
| 276 |
+
'leiden',dendrogram=False,colorbar_title='Expression')
|
| 277 |
+
|
| 278 |
+
|
| 279 |
+
# In[26]:
|
| 280 |
+
|
| 281 |
+
|
| 282 |
+
kidney_lineage_ad.obs['re_anno'] = 'Unknown'
|
| 283 |
+
kidney_lineage_ad.obs.loc[kidney_lineage_ad.obs.leiden.isin(['4']),'re_anno'] = 'Nephron progenitors (E11.5)'
|
| 284 |
+
kidney_lineage_ad.obs.loc[kidney_lineage_ad.obs.leiden.isin(['2','3','1','5']),'re_anno'] = 'Metanephron progenitors (E11.5)'
|
| 285 |
+
kidney_lineage_ad.obs.loc[kidney_lineage_ad.obs.leiden=='0','re_anno'] = 'Kidney (E12.5)'
|
| 286 |
+
|
| 287 |
+
|
| 288 |
+
# In[28]:
|
| 289 |
+
|
| 290 |
+
|
| 291 |
+
# kidney_all = kidney_all[kidney_all.obs.leiden!='3',:]
|
| 292 |
+
kidney_lineage_ad.obs.leiden = list(kidney_lineage_ad.obs.leiden)
|
| 293 |
+
ov.pl.embedding(kidney_lineage_ad,basis='X_umap',
|
| 294 |
+
color=['annotation','re_anno'],
|
| 295 |
+
frameon='small')
|
| 296 |
+
|
| 297 |
+
|
| 298 |
+
# In[29]:
|
| 299 |
+
|
| 300 |
+
|
| 301 |
+
adata1.obs['kidney_anno']=''
|
| 302 |
+
adata1.obs.loc[kidney_lineage_ad[kidney_lineage_ad.obs['week']=='E11.5'].obs.index,'kidney_anno']=kidney_lineage_ad[kidney_lineage_ad.obs['week']=='E11.5'].obs['re_anno']
|
| 303 |
+
|
| 304 |
+
|
| 305 |
+
# In[41]:
|
| 306 |
+
|
| 307 |
+
|
| 308 |
+
import matplotlib.pyplot as plt
|
| 309 |
+
fig, ax = plt.subplots(1, 1, figsize=(8, 8))
|
| 310 |
+
sc.pl.spatial(adata1, color='kidney_anno', spot_size=1.5,
|
| 311 |
+
palette=['#F5F5F5','#ff7f0e', 'green',],show=False,ax=ax)
|
| 312 |
+
|
| 313 |
+
|
| 314 |
+
# We can also differentially analyse Kidney's developmental pedigree to find different marker genes, and we can analyse transcription factors and thus find the regulatory units involved.
|
| 315 |
+
|
| 316 |
+
# In[42]:
|
| 317 |
+
|
| 318 |
+
|
| 319 |
+
test_adata=kidney_lineage_ad
|
| 320 |
+
dds=ov.bulk.pyDEG(test_adata.to_df(layer='lognorm').T)
|
| 321 |
+
dds.drop_duplicates_index()
|
| 322 |
+
print('... drop_duplicates_index success')
|
| 323 |
+
treatment_groups=test_adata.obs[test_adata.obs['week']=='E12.5'].index.tolist()
|
| 324 |
+
control_groups=test_adata.obs[test_adata.obs['week']=='E11.5'].index.tolist()
|
| 325 |
+
result=dds.deg_analysis(treatment_groups,control_groups,method='ttest')
|
| 326 |
+
# -1 means automatically calculates
|
| 327 |
+
dds.foldchange_set(fc_threshold=-1,
|
| 328 |
+
pval_threshold=0.05,
|
| 329 |
+
logp_max=10)
|
| 330 |
+
|
| 331 |
+
|
| 332 |
+
# In[43]:
|
| 333 |
+
|
| 334 |
+
|
| 335 |
+
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
|
| 336 |
+
plot_genes_num=8,plot_genes_fontsize=12,)
|
| 337 |
+
|
| 338 |
+
|
| 339 |
+
# In[52]:
|
| 340 |
+
|
| 341 |
+
|
| 342 |
+
up_gene=dds.result.loc[dds.result['sig']=='up'].sort_values('qvalue')[:3].index.tolist()
|
| 343 |
+
down_gene=dds.result.loc[dds.result['sig']=='down'].sort_values('qvalue')[:3].index.tolist()
|
| 344 |
+
deg_gene=up_gene+down_gene
|
| 345 |
+
|
| 346 |
+
|
| 347 |
+
# In[53]:
|
| 348 |
+
|
| 349 |
+
|
| 350 |
+
sc.pl.dotplot(kidney_lineage_ad,deg_gene,
|
| 351 |
+
groupby='re_anno')
|
| 352 |
+
|
| 353 |
+
|
| 354 |
+
# In addition to analysing directly using differential expression, we can also look for weekly marker genes by considering weeks as categories.
|
| 355 |
+
|
| 356 |
+
# In[55]:
|
| 357 |
+
|
| 358 |
+
|
| 359 |
+
sc.tl.dendrogram(kidney_lineage_ad,'re_anno',use_rep='scaled|original|X_pca')
|
| 360 |
+
sc.tl.rank_genes_groups(kidney_lineage_ad, 're_anno', use_rep='scaled|original|X_pca',
|
| 361 |
+
method='t-test',use_raw=False,key_added='re_anno_ttest')
|
| 362 |
+
sc.pl.rank_genes_groups_dotplot(kidney_lineage_ad,groupby='re_anno',
|
| 363 |
+
cmap='RdBu_r',key='re_anno_ttest',
|
| 364 |
+
standard_scale='var',n_genes=3)
|
| 365 |
+
|
ovrawm/t_spaceflow.txt
ADDED
|
@@ -0,0 +1,160 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Identifying Pseudo-Spatial Map
|
| 5 |
+
#
|
| 6 |
+
# SpaceFlow is Python package for identifying spatiotemporal patterns and spatial domains from Spatial Transcriptomic (ST) Data. Based on deep graph network, SpaceFlow provides the following functions:
|
| 7 |
+
# 1. Encodes the ST data into **low-dimensional embeddings** that reflecting both expression similarity and the spatial proximity of cells in ST data.
|
| 8 |
+
# 2. Incorporates **spatiotemporal** relationships of cells or spots in ST data through a **pseudo-Spatiotemporal Map (pSM)** derived from the embeddings.
|
| 9 |
+
# 3. Identifies **spatial domains** with spatially-coherent expression patterns.
|
| 10 |
+
#
|
| 11 |
+
# Check out [(Ren et al., Nature Communications, 2022)](https://www.nature.com/articles/s41467-022-31739-w) for the detailed methods and applications.
|
| 12 |
+
#
|
| 13 |
+
#
|
| 14 |
+
# 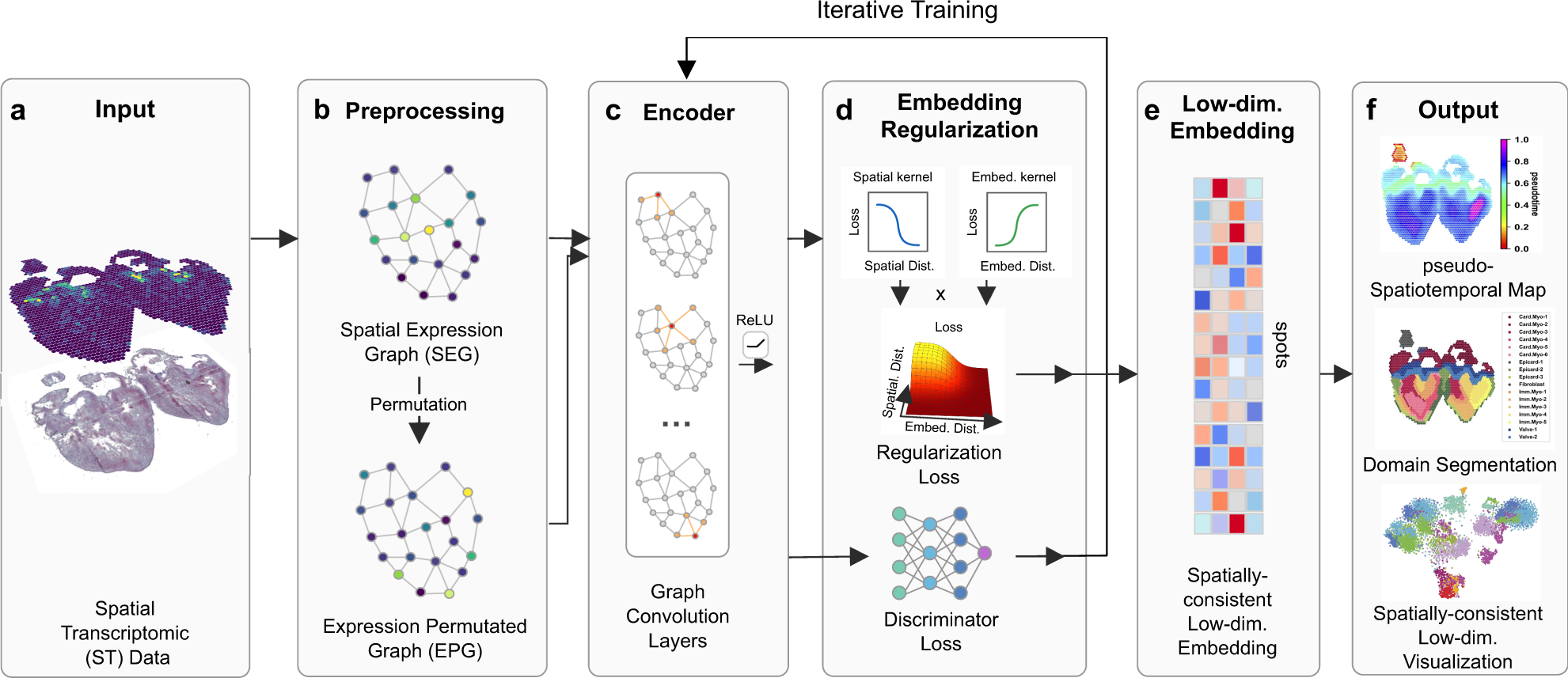
|
| 15 |
+
#
|
| 16 |
+
|
| 17 |
+
# In[1]:
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
import omicverse as ov
|
| 21 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 22 |
+
import scanpy as sc
|
| 23 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 24 |
+
ov.utils.ov_plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# ## Preprocess data
|
| 28 |
+
#
|
| 29 |
+
# Here we present our re-analysis of 151676 sample of the dorsolateral prefrontal cortex (DLPFC) dataset. Maynard et al. has manually annotated DLPFC layers and white matter (WM) based on the morphological features and gene markers.
|
| 30 |
+
#
|
| 31 |
+
# This tutorial demonstrates how to identify spatial domains on 10x Visium data using STAGATE. The processed data are available at https://github.com/LieberInstitute/spatialLIBD. We downloaded the manual annotation from the spatialLIBD package and provided at https://drive.google.com/drive/folders/10lhz5VY7YfvHrtV40MwaqLmWz56U9eBP?usp=sharing.
|
| 32 |
+
|
| 33 |
+
# In[2]:
|
| 34 |
+
|
| 35 |
+
|
| 36 |
+
adata = sc.read_visium(path='data', count_file='151676_filtered_feature_bc_matrix.h5')
|
| 37 |
+
adata.var_names_make_unique()
|
| 38 |
+
|
| 39 |
+
|
| 40 |
+
# <div class="admonition warning">
|
| 41 |
+
# <p class="admonition-title">Note</p>
|
| 42 |
+
# <p>
|
| 43 |
+
# We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code.
|
| 44 |
+
# </p>
|
| 45 |
+
# </div>
|
| 46 |
+
#
|
| 47 |
+
# ```python
|
| 48 |
+
# #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 49 |
+
# #adata.raw = adata
|
| 50 |
+
# #adata = adata[:, adata.var.highly_variable_features]
|
| 51 |
+
# ```
|
| 52 |
+
|
| 53 |
+
# In[3]:
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
sc.pp.calculate_qc_metrics(adata, inplace=True)
|
| 57 |
+
adata = adata[:,adata.var['total_counts']>100]
|
| 58 |
+
adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",)
|
| 59 |
+
adata.raw = adata
|
| 60 |
+
adata = adata[:, adata.var.space_variable_features]
|
| 61 |
+
adata
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# We read the ground truth area of our spatial data
|
| 65 |
+
|
| 66 |
+
# In[4]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
# read the annotation
|
| 70 |
+
import pandas as pd
|
| 71 |
+
import os
|
| 72 |
+
Ann_df = pd.read_csv(os.path.join('data', '151676_truth.txt'), sep='\t', header=None, index_col=0)
|
| 73 |
+
Ann_df.columns = ['Ground Truth']
|
| 74 |
+
adata.obs['Ground Truth'] = Ann_df.loc[adata.obs_names, 'Ground Truth']
|
| 75 |
+
sc.pl.spatial(adata, img_key="hires", color=["Ground Truth"])
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# ## Training the SpaceFlow Model
|
| 79 |
+
#
|
| 80 |
+
# Here, we used `ov.space.pySpaceFlow` to construct a SpaceFlow Object and train the model.
|
| 81 |
+
#
|
| 82 |
+
# We need to store the space location info in `adata.obsm['spatial']`
|
| 83 |
+
|
| 84 |
+
# In[5]:
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
sf_obj=ov.space.pySpaceFlow(adata)
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
# We then train a spatially regularized deep graph network model to learn a low-dimensional embedding that reflecting both expression similarity and the spatial proximity of cells in ST data.
|
| 91 |
+
#
|
| 92 |
+
# Parameters:
|
| 93 |
+
# - `spatial_regularization_strength`: the strength of spatial regularization, the larger the more of the spatial coherence in the identified spatial domains and spatiotemporal patterns. (default: 0.1)
|
| 94 |
+
# - `z_dim`: the target size of the learned embedding. (default: 50)
|
| 95 |
+
# - `lr`: learning rate for optimizing the model. (default: 1e-3)
|
| 96 |
+
# - `epochs`: the max number of the epochs for model training. (default: 1000)
|
| 97 |
+
# - `max_patience`: the max number of the epoch for waiting the loss decreasing. If loss does not decrease for epochs larger than this threshold, the learning will stop, and the model with the parameters that shows the minimal loss are kept as the best model. (default: 50)
|
| 98 |
+
# - `min_stop`: the earliest epoch the learning can stop if no decrease in loss for epochs larger than the `max_patience`. (default: 100)
|
| 99 |
+
# - `random_seed`: the random seed set to the random generators of the `random`, `numpy`, `torch` packages. (default: 42)
|
| 100 |
+
# - `gpu`: the index of the Nvidia GPU, if no GPU, the model will be trained via CPU, which is slower than the GPU training time. (default: 0)
|
| 101 |
+
# - `regularization_acceleration`: whether or not accelerate the calculation of regularization loss using edge subsetting strategy (default: True)
|
| 102 |
+
# - `edge_subset_sz`: the edge subset size for regularization acceleration (default: 1000000)
|
| 103 |
+
#
|
| 104 |
+
|
| 105 |
+
# In[6]:
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
sf_obj.train(spatial_regularization_strength=0.1,
|
| 109 |
+
z_dim=50, lr=1e-3, epochs=1000,
|
| 110 |
+
max_patience=50, min_stop=100,
|
| 111 |
+
random_seed=42, gpu=0,
|
| 112 |
+
regularization_acceleration=True, edge_subset_sz=1000000)
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
# ## Calculated the Pseudo-Spatial Map
|
| 116 |
+
#
|
| 117 |
+
# Unlike the original SpaceFlow, we only need to use the `cal_PSM` function when calling SpaceFlow in omicverse to compute the pSM.
|
| 118 |
+
|
| 119 |
+
# In[7]:
|
| 120 |
+
|
| 121 |
+
|
| 122 |
+
sf_obj.cal_pSM(n_neighbors=20,resolution=1,
|
| 123 |
+
max_cell_for_subsampling=5000,psm_key='pSM_spaceflow')
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
# In[8]:
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
sc.pl.spatial(adata, color=['pSM_spaceflow','Ground Truth'],cmap='RdBu_r')
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# ## Clustering the space
|
| 133 |
+
#
|
| 134 |
+
# We can use `GMM`, `leiden` or `louvain` to cluster the space.
|
| 135 |
+
#
|
| 136 |
+
# ```python
|
| 137 |
+
# sc.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
|
| 138 |
+
# use_rep='spaceflow')
|
| 139 |
+
# ov.utils.cluster(adata,use_rep='spaceflow',method='louvain',resolution=1)
|
| 140 |
+
# ov.utils.cluster(adata,use_rep='spaceflow',method='leiden',resolution=1)
|
| 141 |
+
# ```
|
| 142 |
+
|
| 143 |
+
# In[9]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
ov.utils.cluster(adata,use_rep='spaceflow',method='GMM',n_components=7,covariance_type='full',
|
| 147 |
+
tol=1e-9, max_iter=1000, random_state=3607)
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
# In[10]:
|
| 151 |
+
|
| 152 |
+
|
| 153 |
+
sc.pl.spatial(adata, color=['gmm_cluster',"Ground Truth"])
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# In[ ]:
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
|
ovrawm/t_stagate.txt
ADDED
|
@@ -0,0 +1,296 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Spatial clustering and denoising expressions
|
| 5 |
+
#
|
| 6 |
+
# Spatial clustering, which shares an analogy with single-cell clustering, has expanded the scope of tissue physiology studies from cell-centroid to structure-centroid with spatially resolved transcriptomics (SRT) data.
|
| 7 |
+
#
|
| 8 |
+
# Here, we presented two spatial clustering methods in OmicVerse.
|
| 9 |
+
#
|
| 10 |
+
# We made three improvements in integrating the `GraphST` and `STAGATE` algorithm in OmicVerse:
|
| 11 |
+
# - We removed the preprocessing that comes with `GraphST` and used the preprocessing consistent with all SRTs in OmicVerse
|
| 12 |
+
# - We optimised the dimensional display of `GraphST`, and PCA is considered a self-contained computational step.
|
| 13 |
+
# - We implemented `mclust` using Python, removing the R language dependency.
|
| 14 |
+
#
|
| 15 |
+
# If you found this tutorial helpful, please cite `GraphST`, `STAGATE` and OmicVerse:
|
| 16 |
+
#
|
| 17 |
+
# - Long, Y., Ang, K.S., Li, M. et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat Commun 14, 1155 (2023). https://doi.org/10.1038/s41467-023-36796-3
|
| 18 |
+
# - Dong, K., Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 13, 1739 (2022). https://doi.org/10.1038/s41467-022-29439-6
|
| 19 |
+
#
|
| 20 |
+
#
|
| 21 |
+
|
| 22 |
+
# In[1]:
|
| 23 |
+
|
| 24 |
+
|
| 25 |
+
import omicverse as ov
|
| 26 |
+
#print(f"omicverse version: {ov.__version__}")
|
| 27 |
+
import scanpy as sc
|
| 28 |
+
#print(f"scanpy version: {sc.__version__}")
|
| 29 |
+
ov.plot_set()
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
# ## Preprocess data
|
| 33 |
+
#
|
| 34 |
+
# Here we present our re-analysis of 151676 sample of the dorsolateral prefrontal cortex (DLPFC) dataset. Maynard et al. has manually annotated DLPFC layers and white matter (WM) based on the morphological features and gene markers.
|
| 35 |
+
#
|
| 36 |
+
# This tutorial demonstrates how to identify spatial domains on 10x Visium data using STAGATE. The processed data are available at https://github.com/LieberInstitute/spatialLIBD. We downloaded the manual annotation from the spatialLIBD package and provided at https://drive.google.com/drive/folders/10lhz5VY7YfvHrtV40MwaqLmWz56U9eBP?usp=sharing.
|
| 37 |
+
|
| 38 |
+
# In[2]:
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
adata = sc.read_visium(path='data', count_file='151676_filtered_feature_bc_matrix.h5')
|
| 42 |
+
adata.var_names_make_unique()
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# <div class="admonition warning">
|
| 46 |
+
# <p class="admonition-title">Note</p>
|
| 47 |
+
# <p>
|
| 48 |
+
# We introduced the spatial special svg calculation module prost in omicverse versions greater than `1.6.0` to replace scanpy's HVGs, if you want to use scanpy's HVGs you can set mode=`scanpy` in `ov.space.svg` or use the following code.
|
| 49 |
+
# </p>
|
| 50 |
+
# </div>
|
| 51 |
+
#
|
| 52 |
+
# ```python
|
| 53 |
+
# #adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,target_sum=1e4)
|
| 54 |
+
# #adata.raw = adata
|
| 55 |
+
# #adata = adata[:, adata.var.highly_variable_features]
|
| 56 |
+
# ```
|
| 57 |
+
|
| 58 |
+
# In[4]:
|
| 59 |
+
|
| 60 |
+
|
| 61 |
+
sc.pp.calculate_qc_metrics(adata, inplace=True)
|
| 62 |
+
adata = adata[:,adata.var['total_counts']>100]
|
| 63 |
+
adata=ov.space.svg(adata,mode='prost',n_svgs=3000,target_sum=1e4,platform="visium",)
|
| 64 |
+
adata
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# In[5]:
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
adata.write('data/cluster_svg.h5ad',compression='gzip')
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# In[2]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
#adata=ov.read('data/cluster_svg.h5ad',compression='gzip')
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# (Optional) We read the ground truth area of our spatial data
|
| 80 |
+
#
|
| 81 |
+
# This step is not mandatory to run, in the tutorial, it's just to demonstrate the accuracy of our clustering effect, and in your own tasks, there is often no Ground_truth
|
| 82 |
+
|
| 83 |
+
# In[3]:
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
# read the annotation
|
| 87 |
+
import pandas as pd
|
| 88 |
+
import os
|
| 89 |
+
Ann_df = pd.read_csv(os.path.join('data', '151676_truth.txt'), sep='\t', header=None, index_col=0)
|
| 90 |
+
Ann_df.columns = ['Ground Truth']
|
| 91 |
+
adata.obs['Ground Truth'] = Ann_df.loc[adata.obs_names, 'Ground Truth']
|
| 92 |
+
sc.pl.spatial(adata, img_key="hires", color=["Ground Truth"])
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
# ## GraphST model
|
| 96 |
+
#
|
| 97 |
+
# GraphST was rated as one of the best spatial clustering algorithms on Nature Method 2024.04, so we first tried to call GraphST for spatial domain identification in OmicVerse.
|
| 98 |
+
|
| 99 |
+
# In[64]:
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
# define model
|
| 103 |
+
model = ov.externel.GraphST.GraphST(adata, device='cuda:0')
|
| 104 |
+
|
| 105 |
+
# train model
|
| 106 |
+
adata = model.train(n_pcs=30)
|
| 107 |
+
|
| 108 |
+
|
| 109 |
+
# ### Clustering the space
|
| 110 |
+
#
|
| 111 |
+
# We can use `mclust`, `leiden` or `louvain` to cluster the space.
|
| 112 |
+
#
|
| 113 |
+
# Note that we also add optimal transport to optimise the distribution of labels, using `refine_label` for that processing.
|
| 114 |
+
#
|
| 115 |
+
|
| 116 |
+
# In[84]:
|
| 117 |
+
|
| 118 |
+
|
| 119 |
+
ov.utils.cluster(adata,use_rep='graphst|original|X_pca',method='mclust',n_components=10,
|
| 120 |
+
modelNames='EEV', random_state=112,
|
| 121 |
+
)
|
| 122 |
+
adata.obs['mclust_GraphST'] = ov.utils.refine_label(adata, radius=50, key='mclust')
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
# In[87]:
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=20,
|
| 129 |
+
use_rep='graphst|original|X_pca')
|
| 130 |
+
ov.utils.cluster(adata,use_rep='graphst|original|X_pca',method='louvain',resolution=0.7)
|
| 131 |
+
ov.utils.cluster(adata,use_rep='graphst|original|X_pca',method='leiden',resolution=0.7)
|
| 132 |
+
adata.obs['louvain_GraphST'] = ov.utils.refine_label(adata, radius=50, key='louvain')
|
| 133 |
+
adata.obs['leiden_GraphST'] = ov.utils.refine_label(adata, radius=50, key='leiden')
|
| 134 |
+
|
| 135 |
+
|
| 136 |
+
# In[88]:
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
sc.pl.spatial(adata, color=['mclust_GraphST','leiden_GraphST',
|
| 140 |
+
'louvain_GraphST',"Ground Truth"])
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
# <div class="admonition warning">
|
| 144 |
+
# <p class="admonition-title">Note</p>
|
| 145 |
+
# <p>
|
| 146 |
+
# If you find that the clustering is mediocre, you might consider re-running `model.train()`. Why the results improve
|
| 147 |
+
# </p>
|
| 148 |
+
# </div>
|
| 149 |
+
#
|
| 150 |
+
#
|
| 151 |
+
|
| 152 |
+
# ## STAGATE model
|
| 153 |
+
#
|
| 154 |
+
# STAGATE is designed for spatial clustering and denoising expressions of spatial resolved transcriptomics (ST) data.
|
| 155 |
+
#
|
| 156 |
+
# STAGATE learns low-dimensional latent embeddings with both spatial information and gene expressions via a graph attention auto-encoder. The method adopts an attention mechanism in the middle layer of the encoder and decoder, which adaptively learns the edge weights of spatial neighbor networks, and further uses them to update the spot representation by collectively aggregating information from its neighbors. The latent embeddings and the reconstructed expression profiles can be used to downstream tasks such as spatial domain identification, visualization, spatial trajectory inference, data denoising and 3D expression domain extraction.
|
| 157 |
+
#
|
| 158 |
+
# Dong, Kangning, and Shihua Zhang. “Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder.” Nature Communications 13.1 (2022): 1-12.
|
| 159 |
+
#
|
| 160 |
+
#
|
| 161 |
+
# Here, we used `ov.space.pySTAGATE` to construct a STAGATE object to train the model.
|
| 162 |
+
#
|
| 163 |
+
|
| 164 |
+
# In[14]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
#This step sometimes needs to be run twice
|
| 168 |
+
#and you need to check that adata.obs['X'] is correctly assigned instead of NA
|
| 169 |
+
adata.obs['X'] = adata.obsm['spatial'][:,0]
|
| 170 |
+
adata.obs['Y'] = adata.obsm['spatial'][:,1]
|
| 171 |
+
adata.obs['X'][0]
|
| 172 |
+
|
| 173 |
+
|
| 174 |
+
# In[15]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
STA_obj=ov.space.pySTAGATE(adata,num_batch_x=3,num_batch_y=2,
|
| 178 |
+
spatial_key=['X','Y'],rad_cutoff=200,num_epoch = 1000,lr=0.001,
|
| 179 |
+
weight_decay=1e-4,hidden_dims = [512, 30],
|
| 180 |
+
device='cuda:0')
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
# In[16]:
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
STA_obj.train()
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# We stored the latent embedding in `adata.obsm['STAGATE']`, and denoising expression in `adata.layers['STAGATE_ReX']`
|
| 190 |
+
|
| 191 |
+
# In[17]:
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
STA_obj.predicted()
|
| 195 |
+
adata
|
| 196 |
+
|
| 197 |
+
|
| 198 |
+
# ### Clustering the space
|
| 199 |
+
#
|
| 200 |
+
|
| 201 |
+
# In[18]:
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
ov.utils.cluster(adata,use_rep='STAGATE',method='mclust',n_components=8,
|
| 205 |
+
modelNames='EEV', random_state=112,
|
| 206 |
+
)
|
| 207 |
+
adata.obs['mclust_STAGATE'] = ov.utils.refine_label(adata, radius=50, key='mclust')
|
| 208 |
+
|
| 209 |
+
|
| 210 |
+
# In[21]:
|
| 211 |
+
|
| 212 |
+
|
| 213 |
+
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=20,
|
| 214 |
+
use_rep='STAGATE')
|
| 215 |
+
ov.utils.cluster(adata,use_rep='STAGATE',method='louvain',resolution=0.5)
|
| 216 |
+
ov.utils.cluster(adata,use_rep='STAGATE',method='leiden',resolution=0.5)
|
| 217 |
+
adata.obs['louvain_STAGATE'] = ov.utils.refine_label(adata, radius=50, key='louvain')
|
| 218 |
+
adata.obs['leiden_STAGATE'] = ov.utils.refine_label(adata, radius=50, key='leiden')
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
# In[22]:
|
| 222 |
+
|
| 223 |
+
|
| 224 |
+
sc.pl.spatial(adata, color=['mclust_STAGATE','leiden_STAGATE',
|
| 225 |
+
'louvain_STAGATE',"Ground Truth"])
|
| 226 |
+
|
| 227 |
+
|
| 228 |
+
# ### Denoising
|
| 229 |
+
|
| 230 |
+
# In[23]:
|
| 231 |
+
|
| 232 |
+
|
| 233 |
+
adata.var.sort_values('PI',ascending=False).head(10)
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
# In[24]:
|
| 237 |
+
|
| 238 |
+
|
| 239 |
+
plot_gene = 'MBP'
|
| 240 |
+
import matplotlib.pyplot as plt
|
| 241 |
+
fig, axs = plt.subplots(1, 2, figsize=(8, 4))
|
| 242 |
+
sc.pl.spatial(adata, img_key="hires", color=plot_gene, show=False, ax=axs[0], title='RAW_'+plot_gene, vmax='p99')
|
| 243 |
+
sc.pl.spatial(adata, img_key="hires", color=plot_gene, show=False, ax=axs[1], title='STAGATE_'+plot_gene, layer='STAGATE_ReX', vmax='p99')
|
| 244 |
+
|
| 245 |
+
|
| 246 |
+
# ### Calculated the Pseudo-Spatial Map
|
| 247 |
+
#
|
| 248 |
+
# We compared the model results from `SpaceFlow` and `STAGATE`, and to our surprise, STAGATE can also be applied to predict pSM.
|
| 249 |
+
|
| 250 |
+
# In[25]:
|
| 251 |
+
|
| 252 |
+
|
| 253 |
+
STA_obj.cal_pSM(n_neighbors=20,resolution=1,
|
| 254 |
+
max_cell_for_subsampling=5000)
|
| 255 |
+
adata
|
| 256 |
+
|
| 257 |
+
|
| 258 |
+
# In[26]:
|
| 259 |
+
|
| 260 |
+
|
| 261 |
+
sc.pl.spatial(adata, color=['Ground Truth','pSM_STAGATE'],
|
| 262 |
+
cmap='RdBu_r')
|
| 263 |
+
|
| 264 |
+
|
| 265 |
+
# ## Evaluate cluster
|
| 266 |
+
#
|
| 267 |
+
# We use ARI to evaluate the scoring of our clusters against the true values
|
| 268 |
+
#
|
| 269 |
+
|
| 270 |
+
# In[86]:
|
| 271 |
+
|
| 272 |
+
|
| 273 |
+
from sklearn.metrics.cluster import adjusted_rand_score
|
| 274 |
+
|
| 275 |
+
obs_df = adata.obs.dropna()
|
| 276 |
+
#GraphST
|
| 277 |
+
ARI = adjusted_rand_score(obs_df['mclust_GraphST'], obs_df['Ground Truth'])
|
| 278 |
+
print('mclust_GraphST: Adjusted rand index = %.2f' %ARI)
|
| 279 |
+
|
| 280 |
+
ARI = adjusted_rand_score(obs_df['leiden_GraphST'], obs_df['Ground Truth'])
|
| 281 |
+
print('leiden_GraphST: Adjusted rand index = %.2f' %ARI)
|
| 282 |
+
|
| 283 |
+
ARI = adjusted_rand_score(obs_df['louvain_GraphST'], obs_df['Ground Truth'])
|
| 284 |
+
print('louvain_GraphST: Adjusted rand index = %.2f' %ARI)
|
| 285 |
+
|
| 286 |
+
ARI = adjusted_rand_score(obs_df['mclust_STAGATE'], obs_df['Ground Truth'])
|
| 287 |
+
print('mclust_STAGATE: Adjusted rand index = %.2f' %ARI)
|
| 288 |
+
|
| 289 |
+
ARI = adjusted_rand_score(obs_df['leiden_STAGATE'], obs_df['Ground Truth'])
|
| 290 |
+
print('leiden_STAGATE: Adjusted rand index = %.2f' %ARI)
|
| 291 |
+
|
| 292 |
+
ARI = adjusted_rand_score(obs_df['louvain_STAGATE'], obs_df['Ground Truth'])
|
| 293 |
+
print('louvain_STAGATE: Adjusted rand index = %.2f' %ARI)
|
| 294 |
+
|
| 295 |
+
|
| 296 |
+
# It seems that STAGATE outperforms GraphST on this task, but OmicVerse only provides a unified implementation of the algorithm and does not do a full benchmark of the algorithm.
|
ovrawm/t_staligner.txt
ADDED
|
@@ -0,0 +1,155 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Spatial integration and clustering
|
| 5 |
+
#
|
| 6 |
+
# STAligner is designed for alignment and integration of spatially resolved transcriptomics data.
|
| 7 |
+
#
|
| 8 |
+
# STAligner first normalizes the expression profiles for all spots and constructs a spatial neighbor network using the spatial coordinates. STAligner further employs a graph attention auto-encoder neural network to extract spatially aware embedding, and constructs the spot triplets based on current embeddings to guide the alignment process by attracting similar spots and discriminating dissimilar spots across slices. STAligner introduces the triplet loss to update the spot embedding to reduce the distance from the anchor to positive spot, and increase the distance from the anchor to negative spot. The triplet construction and auto-encoder training are optimized iteratively until batch-corrected embeddings are generated. STAligner can be applied to integrate ST datasets to achieve alignment and simultaneous identification of spatial domains from different biological samples in (a), technological platforms (I), developmental (embryonic) stages (II), disease conditions (III) and consecutive slices of a tissue for 3D slice alignment (IV
|
| 9 |
+
#
|
| 10 |
+
# Zhou, X., Dong, K. & Zhang, S. Integrating spatial transcriptomics data across different conditions, technologies and developmental stages. Nat Comput Sci 3, 894–906 (2023). https://doi.org/10.1038/s43588-023-00528-w
|
| 11 |
+
#
|
| 12 |
+
# ).
|
| 13 |
+
|
| 14 |
+
# In[1]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
from scipy.sparse import csr_matrix
|
| 18 |
+
import omicverse as ov
|
| 19 |
+
import scanpy as sc
|
| 20 |
+
import anndata as ad
|
| 21 |
+
import pandas as pd
|
| 22 |
+
import os
|
| 23 |
+
|
| 24 |
+
ov.utils.ov_plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# # Preprocess data
|
| 28 |
+
#
|
| 29 |
+
# Here, We use the mouse olfactory bulb data generated by Stereo-seq and Slide-seqV2. The processed Stereo-seq and Slide-seqV2 data can be downloaded from https://drive.google.com/drive/folders/1Omte1adVFzyRDw7VloOAQYwtv_NjdWcG?usp=share_link. and the original tutorals can be finded from https://staligner.readthedocs.io/en/latest
|
| 30 |
+
#
|
| 31 |
+
# Here is a critical point that must be clarified: for STAligner, it first calculates highly variable genes before concating annadata samples. Therefore, the number of highly variable genes should not be selected too low. Otherwise, in the case of a large number of samples, the downstream features for STAligner training would be insufficient, impacting the model's performance.
|
| 32 |
+
#
|
| 33 |
+
# When using STAligner, it is necessary to adjust the **rad_cutoff** parameter according to different data to ensure that each spot has an **average of 5-10 adjacent spots** connected to it. Such as: "11.3356 neighbors per cell on average."
|
| 34 |
+
#
|
| 35 |
+
|
| 36 |
+
# In[2]:
|
| 37 |
+
|
| 38 |
+
|
| 39 |
+
Batch_list = []
|
| 40 |
+
adj_list = []
|
| 41 |
+
section_ids = ['Slide-seqV2_MoB', 'Stereo-seq_MoB']
|
| 42 |
+
print(section_ids)
|
| 43 |
+
pathway = '/storage/zengjianyangLab/hulei/scRNA-seq/scripts/STAligner'
|
| 44 |
+
|
| 45 |
+
for section_id in section_ids:
|
| 46 |
+
print(section_id)
|
| 47 |
+
adata = sc.read_h5ad(os.path.join(pathway,section_id+".h5ad"))
|
| 48 |
+
|
| 49 |
+
# check whether the adata.X is sparse matrix
|
| 50 |
+
if isinstance(adata.X, pd.DataFrame):
|
| 51 |
+
adata.X = csr_matrix(adata.X)
|
| 52 |
+
else:
|
| 53 |
+
pass
|
| 54 |
+
|
| 55 |
+
adata.var_names_make_unique(join="++")
|
| 56 |
+
|
| 57 |
+
# make spot name unique
|
| 58 |
+
adata.obs_names = [x+'_'+section_id for x in adata.obs_names]
|
| 59 |
+
|
| 60 |
+
# Constructing the spatial network
|
| 61 |
+
ov.space.Cal_Spatial_Net(adata, rad_cutoff=50) # the spatial network are saved in adata.uns[‘adj’]
|
| 62 |
+
|
| 63 |
+
# Normalization
|
| 64 |
+
sc.pp.highly_variable_genes(adata, flavor="seurat_v3", n_top_genes=10000)
|
| 65 |
+
sc.pp.normalize_total(adata, target_sum=1e4)
|
| 66 |
+
sc.pp.log1p(adata)
|
| 67 |
+
|
| 68 |
+
adata = adata[:, adata.var['highly_variable']]
|
| 69 |
+
adj_list.append(adata.uns['adj'])
|
| 70 |
+
Batch_list.append(adata)
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# In[3]:
|
| 74 |
+
|
| 75 |
+
|
| 76 |
+
Batch_list
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# In[4]:
|
| 80 |
+
|
| 81 |
+
|
| 82 |
+
adata_concat = ad.concat(Batch_list, label="slice_name", keys=section_ids)
|
| 83 |
+
adata_concat.obs["batch_name"] = adata_concat.obs["slice_name"].astype('category')
|
| 84 |
+
print('adata_concat.shape: ', adata_concat.shape)
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
# # Training STAligner model
|
| 88 |
+
#
|
| 89 |
+
# Here, we used `ov.space.pySTAligner` to construct a STAGATE object to train the model.
|
| 90 |
+
#
|
| 91 |
+
# We are using the `train_STAligner_subgraph` function from STAligner to reduce GPU memory usage, each slice is considered as a subgraph for training.
|
| 92 |
+
|
| 93 |
+
# In[5]:
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
get_ipython().run_cell_magic('time', '', "# iter_comb is used to specify the order of integration. For example, (0, 1) means slice 0 will be algined with slice 1 as reference.\niter_comb = [(i, i + 1) for i in range(len(section_ids) - 1)]\n\n# Here, to reduce GPU memory usage, each slice is considered as a subgraph for training.\nSTAligner_obj = ov.space.pySTAligner(adata_concat, verbose=True, knn_neigh = 100, n_epochs = 600, iter_comb = iter_comb,\n batch_key = 'batch_name', key_added='STAligner', Batch_list = Batch_list)\n")
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
# In[6]:
|
| 100 |
+
|
| 101 |
+
|
| 102 |
+
STAligner_obj.train()
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
# We stored the latent embedding in `adata.obsm['STAligner']`.
|
| 106 |
+
|
| 107 |
+
# In[7]:
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
adata = STAligner_obj.predicted()
|
| 111 |
+
|
| 112 |
+
|
| 113 |
+
# # Clustering the space
|
| 114 |
+
#
|
| 115 |
+
# We can use `GMM`, `leiden` or `louvain` to cluster the space.
|
| 116 |
+
#
|
| 117 |
+
# `ov.utils.cluster(adata,use_rep='STAligner',method='GMM',n_components=7,covariance_type='full', tol=1e-9, max_iter=1000, random_state=3607`
|
| 118 |
+
#
|
| 119 |
+
# or `sc.pp.neighbors(adata, use_rep='STAligner', random_state=666)`
|
| 120 |
+
# `ov.utils.cluster(adata,use_rSTAlignerGATE',method='leiden',resolution=1)`
|
| 121 |
+
|
| 122 |
+
# In[8]:
|
| 123 |
+
|
| 124 |
+
|
| 125 |
+
sc.pp.neighbors(adata, use_rep='STAligner', random_state=666)
|
| 126 |
+
ov.utils.cluster(adata,use_rep='STAligner',method='leiden',resolution=0.4)
|
| 127 |
+
sc.tl.umap(adata, random_state=666)
|
| 128 |
+
sc.pl.umap(adata, color=['batch_name',"leiden"],wspace=0.5)
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
# We can also map the clustering results back to the original spatial coordinates to obtain spatially specific clustering results.
|
| 132 |
+
|
| 133 |
+
# In[9]:
|
| 134 |
+
|
| 135 |
+
|
| 136 |
+
import matplotlib.pyplot as plt
|
| 137 |
+
spot_size = 50
|
| 138 |
+
title_size = 15
|
| 139 |
+
fig, ax = plt.subplots(1, 2, figsize=(6, 3), gridspec_kw={'wspace': 0.05, 'hspace': 0.2})
|
| 140 |
+
_sc_0 = sc.pl.spatial(adata[adata.obs['batch_name'] == 'Slide-seqV2_MoB'], img_key=None, color=['leiden'], title=['Slide-seqV2'],
|
| 141 |
+
legend_fontsize=10, show=False, ax=ax[0], frameon=False, spot_size=spot_size, legend_loc=None)
|
| 142 |
+
_sc_0[0].set_title('Slide-seqV2', size=title_size)
|
| 143 |
+
|
| 144 |
+
_sc_1 = sc.pl.spatial(adata[adata.obs['batch_name'] == 'Stereo-seq_MoB'], img_key=None, color=['leiden'], title=['Stereo-seq'],
|
| 145 |
+
legend_fontsize=10, show=False, ax=ax[1], frameon=False, spot_size=spot_size)
|
| 146 |
+
_sc_1[0].set_title('Stereo-seq',size=title_size)
|
| 147 |
+
_sc_1[0].invert_yaxis()
|
| 148 |
+
plt.show()
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
# In[ ]:
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
|
ovrawm/t_starfysh.txt
ADDED
|
@@ -0,0 +1,519 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Deconvolution spatial transcriptomic without scRNA-seq
|
| 5 |
+
#
|
| 6 |
+
# This is a tutorial on an example real Spatial Transcriptomics (ST) data (CID44971_TNBC) from Wu et al., 2021. Raw tutorial could be found in https://starfysh.readthedocs.io/en/latest/notebooks/Starfysh_tutorial_real.html
|
| 7 |
+
#
|
| 8 |
+
#
|
| 9 |
+
# Starfysh performs cell-type deconvolution followed by various downstream analyses to discover spatial interactions in tumor microenvironment. Specifically, Starfysh looks for anchor spots (presumably with the highest compositions of one given cell type) informed by user-provided gene signatures ([see example](https://drive.google.com/file/d/1AXWQy_mwzFEKNjAdrJjXuegB3onxJoOM/view?usp=share_link)) as priors to guide the deconvolution inference, which further enables downstream analyses such as sample integration, spatial hub characterization, cell-cell interactions, etc. This tutorial focuses on the deconvolution task. Overall, Starfysh provides the following options:
|
| 10 |
+
#
|
| 11 |
+
# At omicverse, we have made the following improvements:
|
| 12 |
+
# - Easier visualization, you can use omicverse unified visualization for scientific mapping
|
| 13 |
+
# - Reduce installation dependency errors, we optimized the automatic selection of different packages, you don't need to install too many extra packages and cause conflicts.
|
| 14 |
+
#
|
| 15 |
+
# **Base feature**:
|
| 16 |
+
#
|
| 17 |
+
# - Spot-level deconvolution with expected cell types and corresponding annotated signature gene sets (default)
|
| 18 |
+
#
|
| 19 |
+
# **Optional**:
|
| 20 |
+
#
|
| 21 |
+
# - Archetypal Analysis (AA):
|
| 22 |
+
#
|
| 23 |
+
# *If gene signatures are not provided*
|
| 24 |
+
#
|
| 25 |
+
# - Unsupervised cell type annotation
|
| 26 |
+
#
|
| 27 |
+
# *If gene signatures are provided but require refinement*:
|
| 28 |
+
#
|
| 29 |
+
# - Novel cell type / cell state discovery (complementary to known cell types from the *signatures*)
|
| 30 |
+
# - Refine known marker genes by appending archetype-specific differentially expressed genes, and update anchor spots accordingly
|
| 31 |
+
#
|
| 32 |
+
# - Product-of-Experts (PoE) integration
|
| 33 |
+
#
|
| 34 |
+
# Multi-modal integrative predictions with expression & histology image by leverging additional side information (e.g. cell density) from H&E image.
|
| 35 |
+
#
|
| 36 |
+
# He, S., Jin, Y., Nazaret, A. et al.
|
| 37 |
+
# Starfysh integrates spatial transcriptomic and histologic data to reveal heterogeneous tumor–immune hubs.
|
| 38 |
+
# Nat Biotechnol (2024).
|
| 39 |
+
# https://doi.org/10.1038/s41587-024-02173-8
|
| 40 |
+
|
| 41 |
+
# In[1]:
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
import scanpy as sc
|
| 45 |
+
import omicverse as ov
|
| 46 |
+
ov.plot_set()
|
| 47 |
+
|
| 48 |
+
|
| 49 |
+
# In[2]:
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
from omicverse.externel.starfysh import (AA, utils, plot_utils, post_analysis)
|
| 53 |
+
from omicverse.externel.starfysh import _starfysh as sf_model
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
# ### (1). load data and marker genes
|
| 57 |
+
#
|
| 58 |
+
# File Input:
|
| 59 |
+
# - Spatial transcriptomics
|
| 60 |
+
# - Count matrix: `adata`
|
| 61 |
+
# - (Optional): Paired histology & spot coordinates: `img`, `map_info`
|
| 62 |
+
#
|
| 63 |
+
# - Annotated signatures (marker genes) for potential cell types: `gene_sig`
|
| 64 |
+
#
|
| 65 |
+
# Starfysh is built upon scanpy and Anndata. The common ST/Visium data sample folder consists a expression count file (usually `filtered_featyur_bc_matrix.h5`), and a subdirectory with corresponding H&E image and spatial information, as provided by Visium platform.
|
| 66 |
+
#
|
| 67 |
+
# For example, our example real ST data has the following structure:
|
| 68 |
+
# ```
|
| 69 |
+
# ├── data_folder
|
| 70 |
+
# signature.csv
|
| 71 |
+
#
|
| 72 |
+
# ├── CID44971:
|
| 73 |
+
# \__ filtered_feature_bc_mactrix.h5
|
| 74 |
+
#
|
| 75 |
+
# ├── spatial:
|
| 76 |
+
# \__ aligned_fiducials.jpg
|
| 77 |
+
# detected_tissue_image.jpg
|
| 78 |
+
# scalefactors_json.json
|
| 79 |
+
# tissue_hires_image.png
|
| 80 |
+
# tissue_lowres_image.png
|
| 81 |
+
# tissue_positions_list.csv
|
| 82 |
+
# ```
|
| 83 |
+
#
|
| 84 |
+
# For data that doesn't follow the common visium data structure (e.g. missing `filtered_featyur_bc_matrix.h5` or the given `.h5ad` count matrix file lacks spatial metadata), please construct the data as Anndata synthesizing information as the example simulated data shows:
|
| 85 |
+
|
| 86 |
+
# [Note]: If you’re running this tutorial locally, please download the sample [data](https://drive.google.com/drive/folders/1RIp0Z2eF1m8Ortx0sgB4z5g5ISsRFzJ4?usp=share_link) and [signature gene sets](https://drive.google.com/file/d/1AXWQy_mwzFEKNjAdrJjXuegB3onxJoOM/view?usp=share_link), and save it in the relative path `data/star_data` (otherwise please modify the data_path defined in the cell below):
|
| 87 |
+
|
| 88 |
+
# In[3]:
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# Specify data paths
|
| 92 |
+
data_path = 'data/star_data'
|
| 93 |
+
sample_id = 'CID44971_TNBC'
|
| 94 |
+
sig_name = 'bc_signatures_version_1013.csv'
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# In[4]:
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# Load expression counts and signature gene sets
|
| 101 |
+
adata, adata_normed = utils.load_adata(data_folder=data_path,
|
| 102 |
+
sample_id=sample_id, # sample id
|
| 103 |
+
n_genes=2000 # number of highly variable genes to keep
|
| 104 |
+
)
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
# In[5]:
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
import pandas as pd
|
| 111 |
+
import os
|
| 112 |
+
gene_sig = pd.read_csv(os.path.join(data_path, sig_name))
|
| 113 |
+
gene_sig = utils.filter_gene_sig(gene_sig, adata.to_df())
|
| 114 |
+
gene_sig.head()
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
# **If there's no input signature gene sets, Starfysh defines "archetypal marker genes" as *signatures*. Please refer to the following code snippet and see details in section (3).**
|
| 118 |
+
#
|
| 119 |
+
# ```Python
|
| 120 |
+
# aa_model = AA.ArchetypalAnalysis(adata_orig=adata_normed)
|
| 121 |
+
# archetype, arche_dict, major_idx, evs = aa_model.compute_archetypes(r=40)
|
| 122 |
+
# gene_sig = aa_model.find_markers(n_markers=30, display=False)
|
| 123 |
+
# gene_sig = utils.filter_gene_sig(gene_sig, adata.to_df())
|
| 124 |
+
# gene_sig.head()
|
| 125 |
+
# ```
|
| 126 |
+
|
| 127 |
+
# In[6]:
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
# Load spatial information
|
| 131 |
+
img_metadata = utils.preprocess_img(data_path,
|
| 132 |
+
sample_id,
|
| 133 |
+
adata_index=adata.obs.index,
|
| 134 |
+
#hchannel=False
|
| 135 |
+
)
|
| 136 |
+
img, map_info, scalefactor = img_metadata['img'], img_metadata['map_info'], img_metadata['scalefactor']
|
| 137 |
+
umap_df = utils.get_umap(adata, display=True)
|
| 138 |
+
|
| 139 |
+
|
| 140 |
+
# In[7]:
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
import matplotlib.pyplot as plt
|
| 144 |
+
plt.figure(figsize=(6, 6), dpi=80)
|
| 145 |
+
plt.imshow(img)
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
# In[8]:
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
map_info.head()
|
| 152 |
+
|
| 153 |
+
|
| 154 |
+
# ### (2). Preprocessing (finding anchor spots)
|
| 155 |
+
# - Identify anchor spot locations.
|
| 156 |
+
#
|
| 157 |
+
# Instantiate parameters for Starfysh model training:
|
| 158 |
+
# - Raw & normalized counts after taking highly variable genes
|
| 159 |
+
# - filtered signature genes
|
| 160 |
+
# - library size & spatial smoothed library size (log-transformed)
|
| 161 |
+
# - Anchor spot indices (`anchors_df`) for each cell type & their signature means (`sig_means`)
|
| 162 |
+
#
|
| 163 |
+
|
| 164 |
+
# In[ ]:
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# Parameters for training
|
| 168 |
+
visium_args = utils.VisiumArguments(adata,
|
| 169 |
+
adata_normed,
|
| 170 |
+
gene_sig,
|
| 171 |
+
img_metadata,
|
| 172 |
+
n_anchors=60,
|
| 173 |
+
window_size=3,
|
| 174 |
+
sample_id=sample_id
|
| 175 |
+
)
|
| 176 |
+
|
| 177 |
+
adata, adata_normed = visium_args.get_adata()
|
| 178 |
+
anchors_df = visium_args.get_anchors()
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
# In[10]:
|
| 182 |
+
|
| 183 |
+
|
| 184 |
+
adata.obs['log library size']=visium_args.log_lib
|
| 185 |
+
adata.obs['windowed log library size']=visium_args.win_loglib
|
| 186 |
+
|
| 187 |
+
|
| 188 |
+
# In[11]:
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
sc.pl.spatial(adata, cmap='magma',
|
| 192 |
+
# show first 8 cell types
|
| 193 |
+
color='log library size',
|
| 194 |
+
ncols=4, size=1.3,
|
| 195 |
+
img_key='hires',
|
| 196 |
+
#palette=Layer_color
|
| 197 |
+
# limit color scale at 99.2% quantile of cell abundance
|
| 198 |
+
#vmin=0, vmax='p99.2'
|
| 199 |
+
)
|
| 200 |
+
|
| 201 |
+
|
| 202 |
+
# In[12]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
sc.pl.spatial(adata, cmap='magma',
|
| 206 |
+
# show first 8 cell types
|
| 207 |
+
color='windowed log library size',
|
| 208 |
+
ncols=4, size=1.3,
|
| 209 |
+
img_key='hires',
|
| 210 |
+
#palette=Layer_color
|
| 211 |
+
# limit color scale at 99.2% quantile of cell abundance
|
| 212 |
+
#vmin=0, vmax='p99.2'
|
| 213 |
+
)
|
| 214 |
+
|
| 215 |
+
|
| 216 |
+
# plot raw gene expression:
|
| 217 |
+
|
| 218 |
+
# In[13]:
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
sc.pl.spatial(adata, cmap='magma',
|
| 222 |
+
# show first 8 cell types
|
| 223 |
+
color='IL7R',
|
| 224 |
+
ncols=4, size=1.3,
|
| 225 |
+
img_key='hires',
|
| 226 |
+
#palette=Layer_color
|
| 227 |
+
# limit color scale at 99.2% quantile of cell abundance
|
| 228 |
+
#vmin=0, vmax='p99.2'
|
| 229 |
+
)
|
| 230 |
+
|
| 231 |
+
|
| 232 |
+
# In[14]:
|
| 233 |
+
|
| 234 |
+
|
| 235 |
+
plot_utils.plot_anchor_spots(umap_df,
|
| 236 |
+
visium_args.pure_spots,
|
| 237 |
+
visium_args.sig_mean,
|
| 238 |
+
bbox_x=2
|
| 239 |
+
)
|
| 240 |
+
|
| 241 |
+
|
| 242 |
+
# ### (3). Optional: Archetypal Analysis
|
| 243 |
+
# Overview:
|
| 244 |
+
# If users don't provide annotated gene signature sets with cell types, Starfysh identifies candidates for cell types via archetypal analysis (AA). The underlying assumption is that the geometric "extremes" are identified as the purest cell types, whereas all other spots are mixture of the "archetypes". If the users provide the gene signature sets, they can still optionally apply AA to refine marker genes and update anchor spots for known cell types. In addition, AA can identify & assign potential novel cell types / states. Here are the features provided by the optional archetypal analysis:
|
| 245 |
+
# - Finding archetypal spots & assign 1-1 mapping to their closest anchor spot neighbors
|
| 246 |
+
# - Finding archetypal marker genes & append them to marker genes of annotated cell types
|
| 247 |
+
# - Assigning novel cell type / cell states as the most distant archetypes
|
| 248 |
+
#
|
| 249 |
+
# Overall, Starfysh provides the archetypal analysis as a complementary toolkit for automatic cell-type annotation & signature gene completion:<br><br>
|
| 250 |
+
#
|
| 251 |
+
# 1. *If signature genes aren't provided:* <br><br>Archetypal analysis defines the geometric extrema of the data as major cell types with corresponding marker genes.<br><br>
|
| 252 |
+
#
|
| 253 |
+
# 2. *If complete signature genes are known*: <br><br>Users can skip this section and use only the signature priors<br><br>
|
| 254 |
+
#
|
| 255 |
+
# 3. *If signature genes are incomplete or need refinement*: <br><br>Archetypal analysis can be applied to
|
| 256 |
+
# a. Refine signatures of certain cell types
|
| 257 |
+
# b. Find novel cell types / states that haven't been provided from the input signature
|
| 258 |
+
|
| 259 |
+
# #### If signature genes aren't provided
|
| 260 |
+
#
|
| 261 |
+
# Note: <br>
|
| 262 |
+
# - Intrinsic Dimension (ID) estimator is implemented to estimate the lower-bound for the number of archetypes $k$, followed by elbow method with iterations to identify the optimal $k$. By default, a [conditional number](https://scikit-dimension.readthedocs.io/en/latest/skdim.id.FisherS.html) is set as 30; if you believe there are potentially more / fewer cell types, please increase / decrease `cn` accordingly.
|
| 263 |
+
|
| 264 |
+
# Major cell types & corresponding markers are represented by the inferred archetypes:<br><br>
|
| 265 |
+
#
|
| 266 |
+
#
|
| 267 |
+
#
|
| 268 |
+
# ```Python
|
| 269 |
+
# aa_model = AA.ArchetypalAnalysis(adata_orig=adata_normed)
|
| 270 |
+
# archetype, arche_dict, major_idx, evs = aa_model.compute_archetypes(r=40)
|
| 271 |
+
#
|
| 272 |
+
# # (1). Find archetypal spots & archetypal clusters
|
| 273 |
+
# arche_df = aa_model.find_archetypal_spots(major=True)
|
| 274 |
+
#
|
| 275 |
+
# # (2). Define "signature genes" as marker genes associated with each archetypal cluster
|
| 276 |
+
# gene_sig = aa_model.find_markers(n_markers=30, display=False)
|
| 277 |
+
# gene_sig.head()
|
| 278 |
+
# ```
|
| 279 |
+
|
| 280 |
+
# #### If complete signature genes are known
|
| 281 |
+
#
|
| 282 |
+
# Users can skip th section & run Starfysh
|
| 283 |
+
#
|
| 284 |
+
# #### If signature genes are incomplete or require refinement
|
| 285 |
+
#
|
| 286 |
+
# **In this tutorial, we'll show an example of applying best-aligned archetypes to existing `anchors` of given cell type(s) to append signature genes.**
|
| 287 |
+
|
| 288 |
+
# In[ ]:
|
| 289 |
+
|
| 290 |
+
|
| 291 |
+
aa_model = AA.ArchetypalAnalysis(adata_orig=adata_normed)
|
| 292 |
+
archetype, arche_dict, major_idx, evs = aa_model.compute_archetypes(cn=40)
|
| 293 |
+
# (1). Find archetypal spots & archetypal clusters
|
| 294 |
+
arche_df = aa_model.find_archetypal_spots(major=True)
|
| 295 |
+
|
| 296 |
+
# (2). Find marker genes associated with each archetypal cluster
|
| 297 |
+
markers_df = aa_model.find_markers(n_markers=30, display=False)
|
| 298 |
+
|
| 299 |
+
# (3). Map archetypes to closest anchors (1-1 per cell type)
|
| 300 |
+
map_df, map_dict = aa_model.assign_archetypes(anchors_df)
|
| 301 |
+
|
| 302 |
+
# (4). Optional: Find the most distant archetypes that are not assigned to any annotated cell types
|
| 303 |
+
distant_arches = aa_model.find_distant_archetypes(anchors_df, n=3)
|
| 304 |
+
|
| 305 |
+
|
| 306 |
+
# In[16]:
|
| 307 |
+
|
| 308 |
+
|
| 309 |
+
plot_utils.plot_evs(evs, kmin=aa_model.kmin)
|
| 310 |
+
|
| 311 |
+
|
| 312 |
+
# - Visualize archetypes
|
| 313 |
+
|
| 314 |
+
# In[17]:
|
| 315 |
+
|
| 316 |
+
|
| 317 |
+
aa_model.plot_archetypes(do_3d=False, major=True, disp_cluster=False)
|
| 318 |
+
|
| 319 |
+
|
| 320 |
+
# - Visualize archetypal - cell type mapping:
|
| 321 |
+
|
| 322 |
+
# In[18]:
|
| 323 |
+
|
| 324 |
+
|
| 325 |
+
aa_model.plot_mapping(map_df)
|
| 326 |
+
|
| 327 |
+
|
| 328 |
+
# - Application: appending marker genes Append archetypal marker genes with the best-aligned anchors:
|
| 329 |
+
|
| 330 |
+
# In[ ]:
|
| 331 |
+
|
| 332 |
+
|
| 333 |
+
visium_args = utils.refine_anchors(
|
| 334 |
+
visium_args,
|
| 335 |
+
aa_model,
|
| 336 |
+
#thld=0.7, # alignment threshold
|
| 337 |
+
n_genes=5,
|
| 338 |
+
#n_iters=1
|
| 339 |
+
)
|
| 340 |
+
|
| 341 |
+
# Get updated adata & signatures
|
| 342 |
+
adata, adata_normed = visium_args.get_adata()
|
| 343 |
+
gene_sig = visium_args.gene_sig
|
| 344 |
+
cell_types = gene_sig.columns
|
| 345 |
+
|
| 346 |
+
|
| 347 |
+
# ## Run starfysh without histology integration
|
| 348 |
+
#
|
| 349 |
+
#
|
| 350 |
+
#
|
| 351 |
+
# We perform `n_repeat` random restarts and select the best model with lowest loss for parameter `c` (inferred cell-type proportions):
|
| 352 |
+
#
|
| 353 |
+
# ### (1). Model parameters
|
| 354 |
+
|
| 355 |
+
# In[20]:
|
| 356 |
+
|
| 357 |
+
|
| 358 |
+
import torch
|
| 359 |
+
n_repeats = 3
|
| 360 |
+
epochs = 200
|
| 361 |
+
patience = 50
|
| 362 |
+
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
|
| 363 |
+
|
| 364 |
+
|
| 365 |
+
# ### (2). Model training
|
| 366 |
+
#
|
| 367 |
+
# Users can choose to run the one of the following `Starfysh` model without/with histology integration:
|
| 368 |
+
#
|
| 369 |
+
# Without histology integration: setting `utils.run_starfysh(poe=False)` (default)
|
| 370 |
+
#
|
| 371 |
+
# With histology integration: setting `utils.run_starfysh(poe=True)`
|
| 372 |
+
|
| 373 |
+
# In[21]:
|
| 374 |
+
|
| 375 |
+
|
| 376 |
+
# Run models
|
| 377 |
+
model, loss = utils.run_starfysh(visium_args,
|
| 378 |
+
n_repeats=n_repeats,
|
| 379 |
+
epochs=epochs,
|
| 380 |
+
#patience=patience,
|
| 381 |
+
device=device
|
| 382 |
+
)
|
| 383 |
+
|
| 384 |
+
|
| 385 |
+
# ### Downstream analysis
|
| 386 |
+
#
|
| 387 |
+
# ### Parse Starfysh inference output
|
| 388 |
+
|
| 389 |
+
# In[22]:
|
| 390 |
+
|
| 391 |
+
|
| 392 |
+
adata, adata_normed = visium_args.get_adata()
|
| 393 |
+
inference_outputs, generative_outputs,adata_ = sf_model.model_eval(model,
|
| 394 |
+
adata,
|
| 395 |
+
visium_args,
|
| 396 |
+
poe=False,
|
| 397 |
+
device=device)
|
| 398 |
+
|
| 399 |
+
|
| 400 |
+
# ### Visualize starfysh deconvolution results
|
| 401 |
+
#
|
| 402 |
+
# **Gene sig mean vs. inferred prop**
|
| 403 |
+
|
| 404 |
+
# In[31]:
|
| 405 |
+
|
| 406 |
+
|
| 407 |
+
import numpy as np
|
| 408 |
+
n_cell_types = gene_sig.shape[1]
|
| 409 |
+
idx = np.random.randint(0, n_cell_types)
|
| 410 |
+
post_analysis.gene_mean_vs_inferred_prop(inference_outputs,
|
| 411 |
+
visium_args,
|
| 412 |
+
idx=idx,
|
| 413 |
+
figsize=(4,4)
|
| 414 |
+
)
|
| 415 |
+
|
| 416 |
+
|
| 417 |
+
# ### Spatial visualizations:
|
| 418 |
+
#
|
| 419 |
+
# Inferred density on Spatial map:
|
| 420 |
+
|
| 421 |
+
# In[24]:
|
| 422 |
+
|
| 423 |
+
|
| 424 |
+
plot_utils.pl_spatial_inf_feature(adata_, feature='ql_m', cmap='Blues')
|
| 425 |
+
|
| 426 |
+
|
| 427 |
+
# **Inferred cell-type proportions (spatial map):**
|
| 428 |
+
#
|
| 429 |
+
|
| 430 |
+
# In[25]:
|
| 431 |
+
|
| 432 |
+
|
| 433 |
+
def cell2proportion(adata):
|
| 434 |
+
adata_plot=sc.AnnData(adata.X)
|
| 435 |
+
adata_plot.obs=utils.extract_feature(adata_, 'qc_m').obs.copy()
|
| 436 |
+
adata_plot.var=adata.var.copy()
|
| 437 |
+
adata_plot.obsm=adata.obsm.copy()
|
| 438 |
+
adata_plot.obsp=adata.obsp.copy()
|
| 439 |
+
adata_plot.uns=adata.uns.copy()
|
| 440 |
+
return adata_plot
|
| 441 |
+
adata_plot=cell2proportion(adata_)
|
| 442 |
+
|
| 443 |
+
|
| 444 |
+
# In[26]:
|
| 445 |
+
|
| 446 |
+
|
| 447 |
+
adata_plot
|
| 448 |
+
|
| 449 |
+
|
| 450 |
+
# In[27]:
|
| 451 |
+
|
| 452 |
+
|
| 453 |
+
sc.pl.spatial(adata_plot, cmap='Spectral_r',
|
| 454 |
+
# show first 8 cell types
|
| 455 |
+
color=['Basal','LumA','LumB'],
|
| 456 |
+
ncols=4, size=1.3,
|
| 457 |
+
img_key='hires',
|
| 458 |
+
vmin=0, vmax='p90'
|
| 459 |
+
)
|
| 460 |
+
|
| 461 |
+
|
| 462 |
+
# In[28]:
|
| 463 |
+
|
| 464 |
+
|
| 465 |
+
ov.pl.embedding(adata_plot,
|
| 466 |
+
basis='z_umap',
|
| 467 |
+
color=['Basal', 'LumA', 'MBC', 'Normal epithelial'],
|
| 468 |
+
frameon='small',
|
| 469 |
+
vmin=0, vmax='p90',
|
| 470 |
+
cmap='Spectral_r',
|
| 471 |
+
)
|
| 472 |
+
|
| 473 |
+
|
| 474 |
+
# In[29]:
|
| 475 |
+
|
| 476 |
+
|
| 477 |
+
pred_exprs = sf_model.model_ct_exp(model,
|
| 478 |
+
adata,
|
| 479 |
+
visium_args,
|
| 480 |
+
device=device)
|
| 481 |
+
|
| 482 |
+
|
| 483 |
+
# Plot spot-level expression (e.g. `IL7R` from *Effector Memory T cells (Tem)*):
|
| 484 |
+
#
|
| 485 |
+
|
| 486 |
+
# In[30]:
|
| 487 |
+
|
| 488 |
+
|
| 489 |
+
gene='IL7R'
|
| 490 |
+
gene_celltype='Tem'
|
| 491 |
+
adata_.layers[f'infer_{gene_celltype}']=pred_exprs[gene_celltype]
|
| 492 |
+
|
| 493 |
+
sc.pl.spatial(adata_, cmap='Spectral_r',
|
| 494 |
+
# show first 8 cell types
|
| 495 |
+
color=gene,
|
| 496 |
+
title=f'{gene} (Predicted expression)\n{gene_celltype}',
|
| 497 |
+
layer=f'infer_{gene_celltype}',
|
| 498 |
+
ncols=4, size=1.3,
|
| 499 |
+
img_key='hires',
|
| 500 |
+
#vmin=0, vmax='p90'
|
| 501 |
+
)
|
| 502 |
+
|
| 503 |
+
|
| 504 |
+
# ### Save model & inferred parameters
|
| 505 |
+
|
| 506 |
+
# In[ ]:
|
| 507 |
+
|
| 508 |
+
|
| 509 |
+
# Specify output directory
|
| 510 |
+
outdir = './results/'
|
| 511 |
+
if not os.path.exists(outdir):
|
| 512 |
+
os.mkdir(outdir)
|
| 513 |
+
|
| 514 |
+
# save the model
|
| 515 |
+
torch.save(model.state_dict(), os.path.join(outdir, 'starfysh_model.pt'))
|
| 516 |
+
|
| 517 |
+
# save `adata` object with inferred parameters
|
| 518 |
+
adata.write(os.path.join(outdir, 'st.h5ad'))
|
| 519 |
+
|
ovrawm/t_stt.txt
ADDED
|
@@ -0,0 +1,274 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Spatial transition tensor of single cells
|
| 5 |
+
#
|
| 6 |
+
# spatial transition tensor (STT), a method that uses messenger RNA splicing and spatial transcriptomes through a multiscale dynamical model to characterize multistability in space. By learning a four-dimensional transition tensor and spatial-constrained random walk, STT reconstructs cell-state-specific dynamics and spatial state transitions via both short-time local tensor streamlines between cells and long-time transition paths among attractors.
|
| 7 |
+
#
|
| 8 |
+
# We made three improvements in integrating the STT algorithm in OmicVerse:
|
| 9 |
+
#
|
| 10 |
+
# - **More user-friendly function implementation**: we refreshed the unnecessary extra assignments in the original documentation and automated their encapsulation into the `omicverse.space.STT` class.
|
| 11 |
+
# - **Removed version dependencies**: We removed all the strong dependencies such as ``CellRank==1.3.1`` in the original `requierment.txt`, so that users only need to install the OmicVerse package and the latest version of CellRank to make it work perfectly.
|
| 12 |
+
# - **Added clearer function notes**: We have reorganised the unclear areas described in the original tutorial, where you need to go back to the paper to read, in this document.
|
| 13 |
+
#
|
| 14 |
+
# If you found this tutorial helpful, please cite STT and OmicVerse:
|
| 15 |
+
#
|
| 16 |
+
# - Zhou, P., Bocci, F., Li, T. et al. Spatial transition tensor of single cells. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02266-x
|
| 17 |
+
|
| 18 |
+
# In[1]:
|
| 19 |
+
|
| 20 |
+
|
| 21 |
+
import omicverse as ov
|
| 22 |
+
#import omicverse.STT as st
|
| 23 |
+
import scvelo as scv
|
| 24 |
+
import scanpy as sc
|
| 25 |
+
ov.plot_set()
|
| 26 |
+
|
| 27 |
+
|
| 28 |
+
# ## Preprocess data
|
| 29 |
+
#
|
| 30 |
+
# In this tutorial, we focus on demonstrating and reproducing the original author's data, which has been completed with calculations such as `adata.layers[‘Ms’]`, `adata.layers[‘Mu’]`, and so on. And when analysing our own data, we need the following functions to preprocess the raw data
|
| 31 |
+
#
|
| 32 |
+
# ```
|
| 33 |
+
# scv.pp.filter_and_normalise(adata, min_shared_counts=20, n_top_genes=3000)
|
| 34 |
+
# scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
|
| 35 |
+
# ```
|
| 36 |
+
#
|
| 37 |
+
# The `mouse_brain.h5ad` could be found in the [Github:STT](https://github.com/cliffzhou92/STT/tree/release/data)
|
| 38 |
+
|
| 39 |
+
# In[2]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
adata = sc.read_h5ad('mouse_brain.h5ad')
|
| 43 |
+
adata
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
# ## Training STT model
|
| 47 |
+
#
|
| 48 |
+
# Here, we used ov.space.STT to construct a STAGATE object to train the model. We need to set the following parameters during initialisation:
|
| 49 |
+
#
|
| 50 |
+
# - `spatial_loc`: The nulling coordinates for each spot, in 10x genomic data, are typically `adata.obsm[‘spatial’]`, so this parameter is typically set to `spatial`, but here we store it in `xy_loc`.
|
| 51 |
+
# - `region`: This parameter is considered to be the region of the attractor, which we would normally define using spatial annotations or cellular annotation information.
|
| 52 |
+
|
| 53 |
+
# In[3]:
|
| 54 |
+
|
| 55 |
+
|
| 56 |
+
STT_obj=ov.space.STT(adata,spatial_loc='xy_loc',region='Region')
|
| 57 |
+
|
| 58 |
+
|
| 59 |
+
# Note that we need to specify the number of potential attractors first when predicting attractors. In the author's original tutorial and original paper, there is no clear definition for the specification of this parameter. After referring to the author's tutorial, we use the calculated number of leiden of `adata_aggr` as a prediction of the number of potential attractors.
|
| 60 |
+
|
| 61 |
+
# In[4]:
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
STT_obj.stage_estimate()
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
# The authors noted in the original tutorial that a key parameter called ‘spa_weight’ controls the relative weight of the spatial location similarity kernel.
|
| 68 |
+
#
|
| 69 |
+
# Other parameters are further described in the api documentation. Typically `n_stage` is the parameter we are interested in modifying
|
| 70 |
+
|
| 71 |
+
# In[ ]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
STT_obj.train(n_states = 9, n_iter = 15, weight_connectivities = 0.5,
|
| 75 |
+
n_neighbors = 50,thresh_ms_gene = 0.2, spa_weight =0.3)
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
# After the prediction is complete, the attractor is stored in `adata.obs[‘attractor’]`. We can use `ov.pl.embedding` to visualize it.
|
| 79 |
+
|
| 80 |
+
# In[12]:
|
| 81 |
+
|
| 82 |
+
|
| 83 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 84 |
+
color=["attractor"],frameon='small',
|
| 85 |
+
palette=ov.pl.sc_color[11:])
|
| 86 |
+
|
| 87 |
+
|
| 88 |
+
# In[7]:
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 92 |
+
color=["Region"],frameon='small',
|
| 93 |
+
)
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
# ## Pathway analysis
|
| 97 |
+
#
|
| 98 |
+
# In the original tutorial, the author encapsulated the `gseapy==1.0.4` version for access enrichment. Note that the use of this function requires networking, which we have modified so that we can enrich using the local pathway dataset
|
| 99 |
+
#
|
| 100 |
+
# We can download good access data directly in [enrichr](https://maayanlab.cloud/Enrichr/#libraries), such as the `KEGG_2019_mouse` used in this study.
|
| 101 |
+
#
|
| 102 |
+
# https://maayanlab.cloud/Enrichr/geneSetLibrary?mode=text&libraryName=KEGG_2019_Mouse
|
| 103 |
+
|
| 104 |
+
# In[8]:
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
pathway_dict=ov.utils.geneset_prepare('genesets/KEGG_2019_Mouse.txt',organism='Mouse')
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
# In[ ]:
|
| 111 |
+
|
| 112 |
+
|
| 113 |
+
STT_obj.compute_pathway(pathway_dict)
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
# After running the function, we can use the `plot_pathway` function to visualize the similairty between pathway dynamics in the low dimensional embeddings.
|
| 117 |
+
|
| 118 |
+
# In[11]:
|
| 119 |
+
|
| 120 |
+
|
| 121 |
+
fig = STT_obj.plot_pathway(figsize = (10,8),size = 100,fontsize = 12)
|
| 122 |
+
for ax in fig.axes:
|
| 123 |
+
ax.set_xlabel('Embedding 1', fontsize=20) # Adjust font size as needed
|
| 124 |
+
ax.set_ylabel('Embedding 2', fontsize=20) # Adjust font size as needed
|
| 125 |
+
fig.show()
|
| 126 |
+
|
| 127 |
+
|
| 128 |
+
# If we are interested in the specific pathways, we can use the `plot_tensor_pathway` function to visualize the streamlines.
|
| 129 |
+
|
| 130 |
+
# In[13]:
|
| 131 |
+
|
| 132 |
+
|
| 133 |
+
import matplotlib.pyplot as plt
|
| 134 |
+
fig, ax = plt.subplots(1, 1, figsize=(4, 4))
|
| 135 |
+
STT_obj.plot_tensor_pathway(pathway_name = 'Wnt signaling pathway',basis = 'xy_loc',
|
| 136 |
+
ax=ax)
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
# In[14]:
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
fig, ax = plt.subplots(1, 1, figsize=(4, 4))
|
| 143 |
+
STT_obj.plot_tensor_pathway( 'TGF-beta signaling pathway',basis = 'xy_loc',
|
| 144 |
+
ax=ax)
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
# ## Tensor analysis
|
| 148 |
+
#
|
| 149 |
+
# In the author's original paper, a very interesting concept is mentioned, attractor-averaged and attractor-specific tensors.
|
| 150 |
+
#
|
| 151 |
+
# We can analyse the Joint Tensor and thus study the steady state processes of different attractors. If the streamlines are passing through the attractor then the attractor is in a steady state, if the streamlines are emanating/converging from the attractor then the attractor is in a dynamic state.
|
| 152 |
+
#
|
| 153 |
+
# In addition to this, the Unspliced Tensor also reflects the strength as well as the size of the attraction.
|
| 154 |
+
|
| 155 |
+
# In[4]:
|
| 156 |
+
|
| 157 |
+
|
| 158 |
+
STT_obj.plot_tensor(list_attractor = [1,3,5,6],
|
| 159 |
+
filter_cells = True, member_thresh = 0.1, density = 1)
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
# ## Landscape analysis
|
| 163 |
+
#
|
| 164 |
+
# Each attractor corresponds to a spatial steady state, then we can use contour plots to visualise this steady state and use CellRank's correlation function to infer state transitions between different attractors.
|
| 165 |
+
|
| 166 |
+
# In[17]:
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
STT_obj.construct_landscape(coord_key = 'X_xy_loc')
|
| 170 |
+
|
| 171 |
+
|
| 172 |
+
# In[14]:
|
| 173 |
+
|
| 174 |
+
|
| 175 |
+
sc.pl.embedding(adata, color = ['attractor', 'Region'],basis= 'trans_coord')
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
# Method to infer the lineage, either ‘MPFT’(maxium probability flow tree, global) or ‘MPPT’(most probable path tree, local)
|
| 179 |
+
|
| 180 |
+
# In[15]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
STT_obj.infer_lineage(si=3,sf=4, method = 'MPPT',flux_fraction=0.8,color_palette_name = 'tab10',size_point = 8,
|
| 184 |
+
size_text=12)
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
# The Sankey plot displaying the relation between STT attractors (left) and spatial region annotations (right). The width of links indicates the number of cells that share the connected attractor label and region annotation label simultaneously
|
| 188 |
+
|
| 189 |
+
# In[16]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
fig = STT_obj.plot_sankey(adata.obs['attractor'].tolist(),adata.obs['Region'].tolist())
|
| 193 |
+
|
| 194 |
+
|
| 195 |
+
# ## Saving and Loading Data
|
| 196 |
+
#
|
| 197 |
+
# We need to save the data after the calculation is complete, and we provide the load function to load it directly in the next analysis without having to re-analyse it.
|
| 198 |
+
|
| 199 |
+
# In[24]:
|
| 200 |
+
|
| 201 |
+
|
| 202 |
+
#del adata.uns['r2_keep_train']
|
| 203 |
+
#del adata.uns['r2_keep_test']
|
| 204 |
+
#del adata.uns['kernel']
|
| 205 |
+
#del adata.uns['kernel_connectivities']
|
| 206 |
+
|
| 207 |
+
STT_obj.adata.write('data/mouse_brain_adata.h5ad')
|
| 208 |
+
STT_obj.adata_aggr.write('data/mouse_brain_adata_aggr.h5ad')
|
| 209 |
+
|
| 210 |
+
|
| 211 |
+
# In[2]:
|
| 212 |
+
|
| 213 |
+
|
| 214 |
+
adata=ov.read('data/mouse_brain_adata.h5ad')
|
| 215 |
+
adata_aggr=ov.read('data/mouse_brain_adata_aggr.h5ad')
|
| 216 |
+
|
| 217 |
+
|
| 218 |
+
# In[3]:
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
STT_obj=ov.space.STT(adata,spatial_loc='xy_loc',region='Region')
|
| 222 |
+
STT_obj.load(adata,adata_aggr)
|
| 223 |
+
|
| 224 |
+
|
| 225 |
+
# ## Gene Dynamic
|
| 226 |
+
#
|
| 227 |
+
# The genes with high multistability scores possess varying expression levels in both unspliced and spliced counts within various attractors, and show a gradual change during stage transitions. These gene were stored in `adata.var['r2_test']`
|
| 228 |
+
#
|
| 229 |
+
#
|
| 230 |
+
|
| 231 |
+
# In[18]:
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
adata.var['r2_test'].sort_values(ascending=False)
|
| 235 |
+
|
| 236 |
+
|
| 237 |
+
# In[11]:
|
| 238 |
+
|
| 239 |
+
|
| 240 |
+
STT_obj.plot_top_genes(top_genes = 6, ncols = 2, figsize = (8,8),)
|
| 241 |
+
|
| 242 |
+
|
| 243 |
+
# We analysed the attractor 1-related gene Sim1, and we found that in the unspliced Mu matrix (smooth unspliced), Sim1 is expressed low at attractor 1; in the spliced matrix Ms, Sim1 is expressed high at attractor 1. It indicates that there is a dynamic tendency of Sim1 gene at attractor 1, i.e., the direction of Sim1 expression is flowing towards attractor 1.
|
| 244 |
+
#
|
| 245 |
+
# Velo analyses can also illustrate this, although Sim1 expression is not highest at attractor 1.
|
| 246 |
+
|
| 247 |
+
# In[26]:
|
| 248 |
+
|
| 249 |
+
|
| 250 |
+
import matplotlib.pyplot as plt
|
| 251 |
+
fig, axes = plt.subplots(1, 4, figsize=(12, 3))
|
| 252 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 253 |
+
color=["Sim1"],frameon='small',
|
| 254 |
+
title='Sim1:Ms',show=False,
|
| 255 |
+
layer='Ms',cmap='RdBu_r',ax=axes[0]
|
| 256 |
+
)
|
| 257 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 258 |
+
color=["Sim1"],frameon='small',
|
| 259 |
+
title='Sim1:Mu',show=False,
|
| 260 |
+
layer='Mu',cmap='RdBu_r',ax=axes[1]
|
| 261 |
+
)
|
| 262 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 263 |
+
color=["Sim1"],frameon='small',
|
| 264 |
+
title='Sim1:Velo',show=False,
|
| 265 |
+
layer='velo',cmap='RdBu_r',ax=axes[2]
|
| 266 |
+
)
|
| 267 |
+
ov.pl.embedding(adata, basis="xy_loc",
|
| 268 |
+
color=["Sim1"],frameon='small',
|
| 269 |
+
title='Sim1:exp',show=False,
|
| 270 |
+
#layer='Mu',
|
| 271 |
+
cmap='RdBu_r',ax=axes[3]
|
| 272 |
+
)
|
| 273 |
+
plt.tight_layout()
|
| 274 |
+
|
ovrawm/t_tcga.txt
ADDED
|
@@ -0,0 +1,96 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # TCGA database preprocess
|
| 5 |
+
#
|
| 6 |
+
# We often download patient survival data from the TCGA database for analysis in order to verify the importance of genes in cancer. However, the pre-processing of the TCGA database is often a headache. Here, we have introduced the TCGA module in ov, a way to quickly process the file formats we download from the TCGA database. We need to prepare 3 files as input:
|
| 7 |
+
#
|
| 8 |
+
# - gdc_sample_sheet (`.tsv`): The `Sample Sheet` button of TCGA, and we can get `tsv` file from it
|
| 9 |
+
# - gdc_download_files (`folder`): The `Download/Cart` button of TCGA, and we get `tar.gz` included all file you selected/
|
| 10 |
+
# - clinical_cart (`folder`): The `Clinical` button of TCGA, and we can get `tar.gz` included all clinical of your files
|
| 11 |
+
|
| 12 |
+
# In[1]:
|
| 13 |
+
|
| 14 |
+
|
| 15 |
+
import omicverse as ov
|
| 16 |
+
import scanpy as sc
|
| 17 |
+
ov.plot_set()
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
# ## TCGA counts read
|
| 21 |
+
#
|
| 22 |
+
# Here, we use `ov.bulk.TCGA` to perform the `gdc_sample_sheet`, `gdc_download_files` and `clinical_cart` you download before. The raw count, fpkm and tpm matrix will be stored in anndata object
|
| 23 |
+
|
| 24 |
+
# In[2]:
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
get_ipython().run_cell_magic('time', '', "gdc_sample_sheep='data/TCGA_OV/gdc_sample_sheet.2024-07-05.tsv'\ngdc_download_files='data/TCGA_OV/gdc_download_20240705_180129.081531'\nclinical_cart='data/TCGA_OV/clinical.cart.2024-07-05'\naml_tcga=ov.bulk.pyTCGA(gdc_sample_sheep,gdc_download_files,clinical_cart)\naml_tcga.adata_init()\n")
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
# We can save the anndata object for the next use
|
| 31 |
+
|
| 32 |
+
# In[3]:
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
aml_tcga.adata.write_h5ad('data/TCGA_OV/ov_tcga_raw.h5ad',compression='gzip')
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
# Note: Each time we read the anndata file, we need to initialize the TCGA object using three paths so that the subsequent TCGA functions such as survival analysis can be used properly
|
| 39 |
+
#
|
| 40 |
+
# If you wish to create your own TCGA data, we have provided [sample data](https://figshare.com/ndownloader/files/47461946) here for download:
|
| 41 |
+
#
|
| 42 |
+
# TCGA OV: https://figshare.com/ndownloader/files/47461946
|
| 43 |
+
|
| 44 |
+
# In[2]:
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
gdc_sample_sheep='data/TCGA_OV/gdc_sample_sheet.2024-07-05.tsv'
|
| 48 |
+
gdc_download_files='data/TCGA_OV/gdc_download_20240705_180129.081531'
|
| 49 |
+
clinical_cart='data/TCGA_OV/clinical.cart.2024-07-05'
|
| 50 |
+
aml_tcga=ov.bulk.pyTCGA(gdc_sample_sheep,gdc_download_files,clinical_cart)
|
| 51 |
+
aml_tcga.adata_read('data/TCGA_OV/ov_tcga_raw.h5ad')
|
| 52 |
+
|
| 53 |
+
|
| 54 |
+
# ## Meta init
|
| 55 |
+
# As the TCGA reads the gene_id, we need to convert it to gene_name as well as adding basic information about the patient. Therefore we need to initialise the patient's meta information.
|
| 56 |
+
|
| 57 |
+
# In[3]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
aml_tcga.adata_meta_init()
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
# ## Survial init
|
| 64 |
+
# We set up the path for Clinical earlier, but in fact we did not import the patient information in the previous process, we only initially determined the id of the patient's TCGA, so we attracted to initialize the clinical information
|
| 65 |
+
|
| 66 |
+
# In[4]:
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
aml_tcga.survial_init()
|
| 70 |
+
aml_tcga.adata
|
| 71 |
+
|
| 72 |
+
|
| 73 |
+
# To visualize the gene you interested, we can use `survival_analysis` to finish it.
|
| 74 |
+
|
| 75 |
+
# In[5]:
|
| 76 |
+
|
| 77 |
+
|
| 78 |
+
aml_tcga.survival_analysis('MYC',layer='deseq_normalize',plot=True)
|
| 79 |
+
|
| 80 |
+
|
| 81 |
+
# If you want to calculate the survival of all genes, you can also use the `survial_analysis_all` to finish it. It may calculate a lot of times.
|
| 82 |
+
|
| 83 |
+
# In[ ]:
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
aml_tcga.survial_analysis_all()
|
| 87 |
+
aml_tcga.adata
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
# Don't forget to save your result.
|
| 91 |
+
|
| 92 |
+
# In[ ]:
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
aml_tcga.adata.write_h5ad('data/TCGA_OV/ov_tcga_survial_all.h5ad',compression='gzip')
|
| 96 |
+
|
ovrawm/t_tosica.txt
ADDED
|
@@ -0,0 +1,317 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Celltype annotation migration(mapping) with TOSICA
|
| 5 |
+
#
|
| 6 |
+
# We know that when all samples cannot be obtained at the same time, it would be desirable to classify the cell types on the first batch of data and use them to annotate the data obtained later or to be obtained in the future with the same standard, without the need to processing and mapping them together again.
|
| 7 |
+
#
|
| 8 |
+
# So migration(mapping) the reference cell annotation is necessary. This tutorial focuses on how to migration(mapping) the cell annotation from reference scRNA-seq atlas to new scRNA-seq data.
|
| 9 |
+
#
|
| 10 |
+
# Paper: [Transformer for one stop interpretable cell type annotation](https://www.nature.com/articles/s41467-023-35923-4)
|
| 11 |
+
#
|
| 12 |
+
# Code: https://github.com/JackieHanLab/TOSICA
|
| 13 |
+
#
|
| 14 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1BjPEG-kLAgicP8iQvtVtpzzbIOmk1X23?usp=sharing
|
| 15 |
+
#
|
| 16 |
+
# 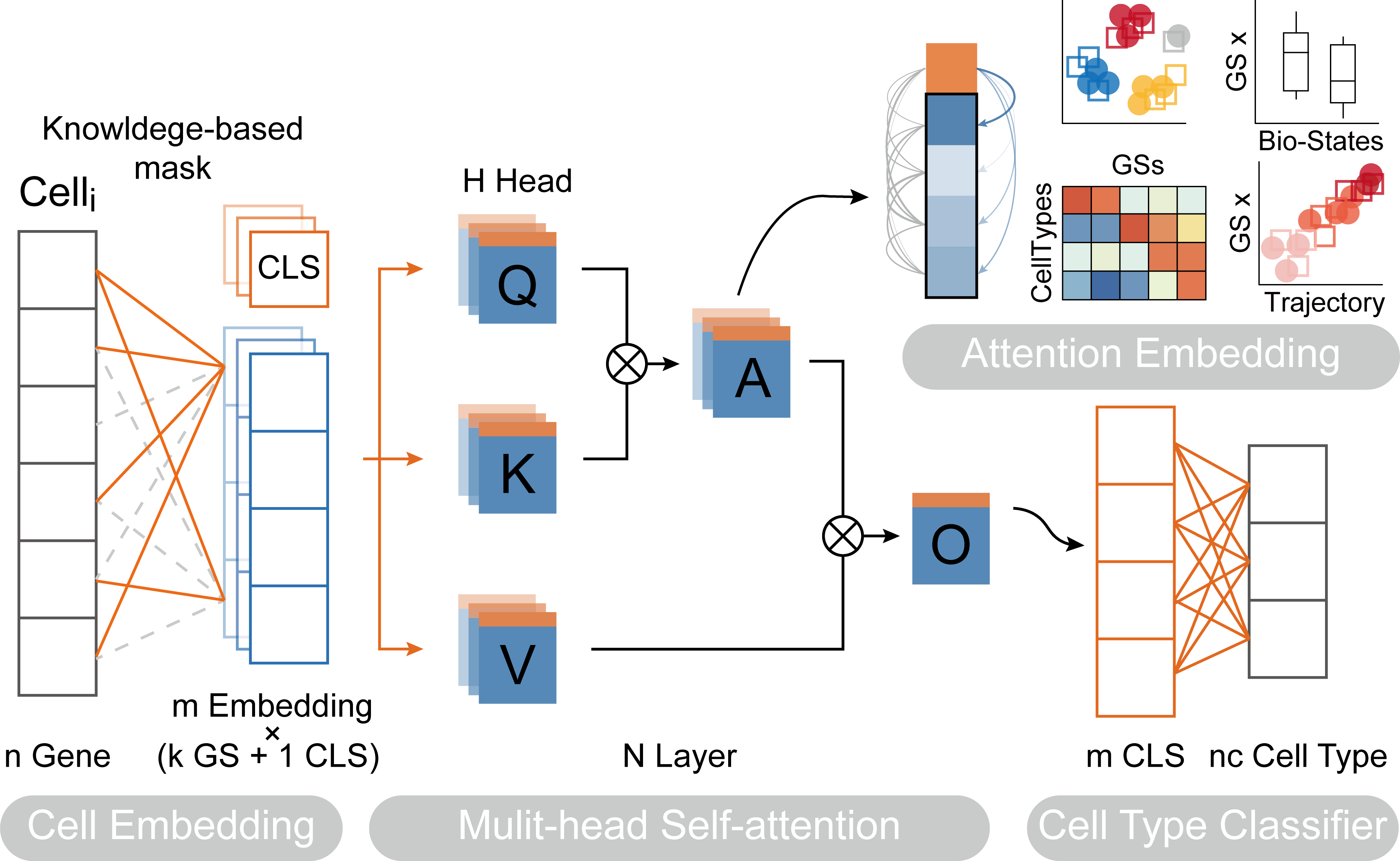
|
| 17 |
+
#
|
| 18 |
+
|
| 19 |
+
# In[1]:
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
import omicverse as ov
|
| 23 |
+
import scanpy as sc
|
| 24 |
+
ov.utils.ov_plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# ## Loading data
|
| 28 |
+
#
|
| 29 |
+
# + `demo_train.h5ad` : Braon(GSE84133) and Muraro(GSE85241)
|
| 30 |
+
#
|
| 31 |
+
# + `demo_test.h5ad` : xin(GSE81608), segerstolpe(E-MTAB-5061) and Lawlor(GSE86473)
|
| 32 |
+
#
|
| 33 |
+
# They can be downloaded at https://figshare.com/projects/TOSICA_demo/158489.
|
| 34 |
+
|
| 35 |
+
# In[2]:
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
ref_adata = sc.read('demo_train.h5ad')
|
| 39 |
+
ref_adata = ref_adata[:,ref_adata.var_names]
|
| 40 |
+
print(ref_adata)
|
| 41 |
+
print(ref_adata.obs.Celltype.value_counts())
|
| 42 |
+
|
| 43 |
+
|
| 44 |
+
# In[4]:
|
| 45 |
+
|
| 46 |
+
|
| 47 |
+
query_adata = sc.read('demo_test.h5ad')
|
| 48 |
+
query_adata = query_adata[:,ref_adata.var_names]
|
| 49 |
+
print(query_adata)
|
| 50 |
+
print(query_adata.obs.Celltype.value_counts())
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
# We need to select the same gene training and predicting the celltype
|
| 54 |
+
|
| 55 |
+
# In[ ]:
|
| 56 |
+
|
| 57 |
+
|
| 58 |
+
ref_adata.var_names_make_unique()
|
| 59 |
+
query_adata.var_names_make_unique()
|
| 60 |
+
ret_gene=list(set(query_adata.var_names) & set(ref_adata.var_names))
|
| 61 |
+
len(ret_gene)
|
| 62 |
+
|
| 63 |
+
|
| 64 |
+
# In[ ]:
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
query_adata=query_adata[:,ret_gene]
|
| 68 |
+
ref_adata=ref_adata[:,ret_gene]
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
# In[5]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
print(f"The max of ref_adata is {ref_adata.X.max()}, query_data is {query_adata.X.max()}",)
|
| 75 |
+
|
| 76 |
+
|
| 77 |
+
# By comparing the maximum values of the two data, we can see that the data has been normalised `sc.pp.normalize_total` and logarithmised `sc.pp.log1p`. The same treatment is applied to the data when we use our own data for analysis.
|
| 78 |
+
#
|
| 79 |
+
# ## Download Genesets
|
| 80 |
+
#
|
| 81 |
+
# Here, we need to download the genesets as pathway at first. You can use `ov.utils.download_tosica_gmt()` to download automatically or manual download from:
|
| 82 |
+
#
|
| 83 |
+
# - 'GO_bp':'https://figshare.com/ndownloader/files/41460072',
|
| 84 |
+
# - 'TF':'https://figshare.com/ndownloader/files/41460066',
|
| 85 |
+
# - 'reactome':'https://figshare.com/ndownloader/files/41460051',
|
| 86 |
+
# - 'm_GO_bp':'https://figshare.com/ndownloader/files/41460060',
|
| 87 |
+
# - 'm_TF':'https://figshare.com/ndownloader/files/41460057',
|
| 88 |
+
# - 'm_reactome':'https://figshare.com/ndownloader/files/41460054',
|
| 89 |
+
# - 'immune':'https://figshare.com/ndownloader/files/41460063',
|
| 90 |
+
#
|
| 91 |
+
|
| 92 |
+
# In[ ]:
|
| 93 |
+
|
| 94 |
+
|
| 95 |
+
ov.utils.download_tosica_gmt()
|
| 96 |
+
|
| 97 |
+
|
| 98 |
+
# ## Initialisation the TOSICA model
|
| 99 |
+
#
|
| 100 |
+
# We first need to train the TOSICA model on the REFERENCE dataset, where omicverse provides a simple class `pyTOSICA`, and all subsequent operations can be done with `pyTOSICA`. We need to set the parameters for model initialisation.
|
| 101 |
+
#
|
| 102 |
+
# - `adata`: the reference adata object
|
| 103 |
+
# - `gmt_path`: default pre-prepared mask or path to .gmt files. you can use `ov.utils.download_tosica_gmt()` to obtain the genesets
|
| 104 |
+
# - `depth`: the depth of transformer model, When it is set to 2, a memory leak may occur
|
| 105 |
+
# - `label_name`: the reference key of celltype in `adata.obs`
|
| 106 |
+
# - `project_path`: the save path of TOSICA model
|
| 107 |
+
# - `batch_size`: indicates the number of cells passed to the programme for training in a single pass
|
| 108 |
+
|
| 109 |
+
# In[6]:
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
tosica_obj=ov.single.pyTOSICA(adata=ref_adata,
|
| 113 |
+
gmt_path='genesets/GO_bp.gmt', depth=1,
|
| 114 |
+
label_name='Celltype',
|
| 115 |
+
project_path='hGOBP_demo',
|
| 116 |
+
batch_size=8)
|
| 117 |
+
|
| 118 |
+
|
| 119 |
+
# ## Training the TOSICA model
|
| 120 |
+
#
|
| 121 |
+
# There're 4 arguments to set when training the TOSICA model.
|
| 122 |
+
#
|
| 123 |
+
# - pre_weights: The path of the pre-trained weights.
|
| 124 |
+
# - lr: The learning rate.
|
| 125 |
+
# - epochs: The number of epochs.
|
| 126 |
+
# - lrf: The learning rate of the last layer.
|
| 127 |
+
|
| 128 |
+
# In[5]:
|
| 129 |
+
|
| 130 |
+
|
| 131 |
+
tosica_obj.train(epochs=5)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# We can use `.save` to store the `TOSICA` model in `project_path`
|
| 135 |
+
|
| 136 |
+
# In[6]:
|
| 137 |
+
|
| 138 |
+
|
| 139 |
+
tosica_obj.save()
|
| 140 |
+
|
| 141 |
+
|
| 142 |
+
# The model can be loaded from `project_path` automatically.
|
| 143 |
+
|
| 144 |
+
# In[7]:
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
tosica_obj.load()
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
# ## Update with query
|
| 151 |
+
#
|
| 152 |
+
|
| 153 |
+
# In[8]:
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
new_adata=tosica_obj.predicted(pre_adata=query_adata)
|
| 157 |
+
|
| 158 |
+
|
| 159 |
+
# ## Visualize the reference and mapping
|
| 160 |
+
#
|
| 161 |
+
# We first compute the lower dimensional space of query_data, where we use omicverse's preprocessing method as well as the mde method for dimensionality reduction
|
| 162 |
+
#
|
| 163 |
+
# To visualize the PCA’s embeddings, we use the `pymde` package wrapper in omicverse. This is an alternative to UMAP that is GPU-accelerated.
|
| 164 |
+
|
| 165 |
+
# In[15]:
|
| 166 |
+
|
| 167 |
+
|
| 168 |
+
ov.pp.scale(query_adata)
|
| 169 |
+
ov.pp.pca(query_adata,layer='scaled',n_pcs=50)
|
| 170 |
+
sc.pp.neighbors(query_adata, n_neighbors=15, n_pcs=50,
|
| 171 |
+
use_rep='scaled|original|X_pca')
|
| 172 |
+
query_adata.obsm["X_mde"] = ov.utils.mde(query_adata.obsm["scaled|original|X_pca"])
|
| 173 |
+
query_adata
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
# Since new_adata and query_adata have the same cells, their low-dimensional spaces are also the same. So we proceed directly to the assignment operation.
|
| 177 |
+
|
| 178 |
+
# In[16]:
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
new_adata.obsm=query_adata[new_adata.obs.index].obsm.copy()
|
| 182 |
+
new_adata.obsp=query_adata[new_adata.obs.index].obsp.copy()
|
| 183 |
+
new_adata
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
# Since the predicted cell types are not exactly the same as the original cell types, the colours are not exactly the same. For the visualisation effect, we manually set the colour of the predicted cell type with the original cell type.
|
| 187 |
+
|
| 188 |
+
# In[18]:
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
import numpy as np
|
| 192 |
+
col = np.array([
|
| 193 |
+
"#98DF8A","#E41A1C" ,"#377EB8", "#4DAF4A" ,"#984EA3" ,"#FF7F00" ,"#FFFF33" ,"#A65628" ,"#F781BF" ,"#999999","#1F77B4","#FF7F0E","#279E68","#FF9896"
|
| 194 |
+
]).astype('<U7')
|
| 195 |
+
|
| 196 |
+
celltype = ("alpha","beta","ductal","acinar","delta","PP","PSC","endothelial","epsilon","mast","macrophage","schwann",'t_cell')
|
| 197 |
+
new_adata.obs['Prediction'] = new_adata.obs['Prediction'].astype('category')
|
| 198 |
+
new_adata.obs['Prediction'] = new_adata.obs['Prediction'].cat.reorder_categories(list(celltype))
|
| 199 |
+
new_adata.uns['Prediction_colors'] = col[1:]
|
| 200 |
+
|
| 201 |
+
celltype = ("MHC class II","alpha","beta","ductal","acinar","delta","PP","PSC","endothelial","epsilon","mast")
|
| 202 |
+
new_adata.obs['Celltype'] = new_adata.obs['Celltype'].astype('category')
|
| 203 |
+
new_adata.obs['Celltype'] = new_adata.obs['Celltype'].cat.reorder_categories(list(celltype))
|
| 204 |
+
new_adata.uns['Celltype_colors'] = col[:11]
|
| 205 |
+
|
| 206 |
+
|
| 207 |
+
# In[24]:
|
| 208 |
+
|
| 209 |
+
|
| 210 |
+
sc.pl.embedding(
|
| 211 |
+
new_adata,
|
| 212 |
+
basis="X_mde",
|
| 213 |
+
color=['Celltype', 'Prediction'],
|
| 214 |
+
frameon=False,
|
| 215 |
+
#ncols=1,
|
| 216 |
+
wspace=0.5,
|
| 217 |
+
#palette=ov.utils.pyomic_palette()[11:],
|
| 218 |
+
show=False,
|
| 219 |
+
)
|
| 220 |
+
|
| 221 |
+
|
| 222 |
+
# ## Pathway attention
|
| 223 |
+
#
|
| 224 |
+
# TOSICA has another special feature, which is the ability to computationally use self-attention mechanisms to find pathways associated with cell types. Here we demonstrate the approach of this downstream analysis.
|
| 225 |
+
#
|
| 226 |
+
# We first need to filter out the predicted types of cells with cell counts less than 5.
|
| 227 |
+
|
| 228 |
+
# In[30]:
|
| 229 |
+
|
| 230 |
+
|
| 231 |
+
cell_idx=new_adata.obs['Prediction'].value_counts()[new_adata.obs['Prediction'].value_counts()<5].index
|
| 232 |
+
new_adata=new_adata[~new_adata.obs['Prediction'].isin(cell_idx)]
|
| 233 |
+
|
| 234 |
+
|
| 235 |
+
# We then used `sc.tl.rank_genes_groups` to calculate the differential pathways with the highest attention across cell types. This differential pathway is derived from the gmt genesets used for the previous calculation.
|
| 236 |
+
|
| 237 |
+
# In[31]:
|
| 238 |
+
|
| 239 |
+
|
| 240 |
+
sc.tl.rank_genes_groups(new_adata, 'Prediction', method='wilcoxon')
|
| 241 |
+
|
| 242 |
+
|
| 243 |
+
# In[34]:
|
| 244 |
+
|
| 245 |
+
|
| 246 |
+
sc.pl.rank_genes_groups_dotplot(new_adata,
|
| 247 |
+
n_genes=3,standard_scale='var',)
|
| 248 |
+
|
| 249 |
+
|
| 250 |
+
# If you want to get the cell-specific pathway, you can use `sc.get.rank_genes_groups_df` to get.
|
| 251 |
+
#
|
| 252 |
+
# For example, we would like to obtain the pathway with the highest attention for the cell type `PP`
|
| 253 |
+
|
| 254 |
+
# In[35]:
|
| 255 |
+
|
| 256 |
+
|
| 257 |
+
degs = sc.get.rank_genes_groups_df(new_adata, group='PP', key='rank_genes_groups',
|
| 258 |
+
pval_cutoff=0.05)
|
| 259 |
+
degs.head()
|
| 260 |
+
|
| 261 |
+
|
| 262 |
+
# In[36]:
|
| 263 |
+
|
| 264 |
+
|
| 265 |
+
sc.pl.embedding(
|
| 266 |
+
new_adata,
|
| 267 |
+
basis="X_mde",
|
| 268 |
+
color=['Prediction','GOBP_REGULATION_OF_MUSCLE_SYSTEM_PROCESS'],
|
| 269 |
+
frameon=False,
|
| 270 |
+
#ncols=1,
|
| 271 |
+
wspace=0.5,
|
| 272 |
+
#palette=ov.utils.pyomic_palette()[11:],
|
| 273 |
+
show=False,
|
| 274 |
+
)
|
| 275 |
+
|
| 276 |
+
|
| 277 |
+
# If you call omciverse to complete a TOSICA analysis, don't forget to cite the following literature:
|
| 278 |
+
#
|
| 279 |
+
# ```
|
| 280 |
+
# @article{pmid:36641532,
|
| 281 |
+
# journal = {Nature communications},
|
| 282 |
+
# doi = {10.1038/s41467-023-35923-4},
|
| 283 |
+
# issn = {2041-1723},
|
| 284 |
+
# number = {1},
|
| 285 |
+
# pmid = {36641532},
|
| 286 |
+
# pmcid = {PMC9840170},
|
| 287 |
+
# address = {England},
|
| 288 |
+
# title = {Transformer for one stop interpretable cell type annotation},
|
| 289 |
+
# volume = {14},
|
| 290 |
+
# author = {Chen, Jiawei and Xu, Hao and Tao, Wanyu and Chen, Zhaoxiong and Zhao, Yuxuan and Han, Jing-Dong J},
|
| 291 |
+
# note = {[Online; accessed 2023-07-18]},
|
| 292 |
+
# pages = {223},
|
| 293 |
+
# date = {2023-01-14},
|
| 294 |
+
# year = {2023},
|
| 295 |
+
# month = {1},
|
| 296 |
+
# day = {14},
|
| 297 |
+
# }
|
| 298 |
+
#
|
| 299 |
+
#
|
| 300 |
+
# @misc{doi:10.1101/2023.06.06.543913,
|
| 301 |
+
# doi = {10.1101/2023.06.06.543913},
|
| 302 |
+
# publisher = {Cold Spring Harbor Laboratory},
|
| 303 |
+
# title = {OmicVerse: A single pipeline for exploring the entire transcriptome universe},
|
| 304 |
+
# author = {Zeng, Zehua and Ma, Yuqing and Hu, Lei and Xiong, Yuanyan and Du, Hongwu},
|
| 305 |
+
# note = {[Online; accessed 2023-07-18]},
|
| 306 |
+
# date = {2023-06-08},
|
| 307 |
+
# year = {2023},
|
| 308 |
+
# month = {6},
|
| 309 |
+
# day = {8},
|
| 310 |
+
# }
|
| 311 |
+
# ```
|
| 312 |
+
|
| 313 |
+
# In[ ]:
|
| 314 |
+
|
| 315 |
+
|
| 316 |
+
|
| 317 |
+
|
ovrawm/t_traj.txt
ADDED
|
@@ -0,0 +1,238 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Trajectory Inference with PAGA or Palantir
|
| 5 |
+
#
|
| 6 |
+
# Diffusion maps were introduced by Ronald Coifman and Stephane Lafon, and the underlying idea is to assume that the data are samples from a diffusion process.
|
| 7 |
+
#
|
| 8 |
+
# Palantir is an algorithm to align cells along differentiation trajectories. Palantir models differentiation as a stochastic process where stem cells differentiate to terminally differentiated cells by a series of steps through a low dimensional phenotypic manifold. Palantir effectively captures the continuity in cell states and the stochasticity in cell fate determination.
|
| 9 |
+
#
|
| 10 |
+
# Note that both methods require the input of cells in their initial state, and we will introduce other methods that do not require the input of artificial information, such as pyVIA, in subsequent analyses.
|
| 11 |
+
#
|
| 12 |
+
#
|
| 13 |
+
# ## Preprocess data
|
| 14 |
+
#
|
| 15 |
+
# As an example, we apply differential kinetic analysis to dentate gyrus neurogenesis, which comprises multiple heterogeneous subpopulations.
|
| 16 |
+
|
| 17 |
+
# In[1]:
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
import scanpy as sc
|
| 21 |
+
import scvelo as scv
|
| 22 |
+
import matplotlib.pyplot as plt
|
| 23 |
+
import omicverse as ov
|
| 24 |
+
ov.plot_set()
|
| 25 |
+
|
| 26 |
+
|
| 27 |
+
# In[2]:
|
| 28 |
+
|
| 29 |
+
|
| 30 |
+
import scvelo as scv
|
| 31 |
+
adata=scv.datasets.dentategyrus()
|
| 32 |
+
adata
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# In[3]:
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=3000,)
|
| 39 |
+
adata.raw = adata
|
| 40 |
+
adata = adata[:, adata.var.highly_variable_features]
|
| 41 |
+
ov.pp.scale(adata)
|
| 42 |
+
ov.pp.pca(adata,layer='scaled',n_pcs=50)
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# Let us inspect the contribution of single PCs to the total variance in the data. This gives us information about how many PCs we should consider in order to compute the neighborhood relations of cells. In our experience, often a rough estimate of the number of PCs does fine.
|
| 46 |
+
|
| 47 |
+
# In[4]:
|
| 48 |
+
|
| 49 |
+
|
| 50 |
+
ov.utils.plot_pca_variance_ratio(adata)
|
| 51 |
+
|
| 52 |
+
|
| 53 |
+
# ## Trajectory inference with diffusion map
|
| 54 |
+
#
|
| 55 |
+
# Here, we used `ov.single.TrajInfer` to construct a Trajectory Inference object.
|
| 56 |
+
|
| 57 |
+
# In[5]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
Traj=ov.single.TrajInfer(adata,basis='X_umap',groupby='clusters',
|
| 61 |
+
use_rep='scaled|original|X_pca',n_comps=50,)
|
| 62 |
+
Traj.set_origin_cells('nIPC')
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
# In[6]:
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
Traj.inference(method='diffusion_map')
|
| 69 |
+
|
| 70 |
+
|
| 71 |
+
# In[7]:
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 75 |
+
color=['clusters','dpt_pseudotime'],
|
| 76 |
+
frameon='small',cmap='Reds')
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# PAGA graph abstraction has benchmarked as top-performing method for trajectory inference. It provides a graph-like map of the data topology with weighted edges corresponding to the connectivity between two clusters.
|
| 80 |
+
#
|
| 81 |
+
# Here, PAGA is extended by neighbor directionality.
|
| 82 |
+
|
| 83 |
+
# In[8]:
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
ov.utils.cal_paga(adata,use_time_prior='dpt_pseudotime',vkey='paga',
|
| 87 |
+
groups='clusters')
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
# In[9]:
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
ov.utils.plot_paga(adata,basis='umap', size=50, alpha=.1,title='PAGA LTNN-graph',
|
| 94 |
+
min_edge_width=2, node_size_scale=1.5,show=False,legend_loc=False)
|
| 95 |
+
|
| 96 |
+
|
| 97 |
+
# ## Trajectory inference with Slingshot
|
| 98 |
+
#
|
| 99 |
+
# Provides functions for inferring continuous, branching lineage structures in low-dimensional data. Slingshot was designed to model developmental trajectories in single-cell RNA sequencing data and serve as a component in an analysis pipeline after dimensionality reduction and clustering. It is flexible enough to handle arbitrarily many branching events and allows for the incorporation of prior knowledge through supervised graph construction.
|
| 100 |
+
|
| 101 |
+
# In[10]:
|
| 102 |
+
|
| 103 |
+
|
| 104 |
+
Traj=ov.single.TrajInfer(adata,basis='X_umap',groupby='clusters',
|
| 105 |
+
use_rep='scaled|original|X_pca',n_comps=50)
|
| 106 |
+
Traj.set_origin_cells('nIPC')
|
| 107 |
+
#Traj.set_terminal_cells(["Granule mature","OL","Astrocytes"])
|
| 108 |
+
|
| 109 |
+
|
| 110 |
+
# If you only need the proposed timing and not the lineage of the process, then you can leave the debug_axes parameter unset.
|
| 111 |
+
|
| 112 |
+
# In[ ]:
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
Traj.inference(method='slingshot',num_epochs=1)
|
| 116 |
+
|
| 117 |
+
|
| 118 |
+
# else, you can set `debug_axes` to visualize the lineage
|
| 119 |
+
|
| 120 |
+
# In[13]:
|
| 121 |
+
|
| 122 |
+
|
| 123 |
+
fig, axes = plt.subplots(nrows=2, ncols=2, figsize=(8, 8))
|
| 124 |
+
Traj.inference(method='slingshot',num_epochs=1,debug_axes=axes)
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
# In[14]:
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
ov.utils.embedding(adata,basis='X_umap',
|
| 131 |
+
color=['clusters','slingshot_pseudotime'],
|
| 132 |
+
frameon='small',cmap='Reds')
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
# In[15]:
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
sc.pp.neighbors(adata,use_rep='scaled|original|X_pca')
|
| 139 |
+
ov.utils.cal_paga(adata,use_time_prior='slingshot_pseudotime',vkey='paga',
|
| 140 |
+
groups='clusters')
|
| 141 |
+
|
| 142 |
+
|
| 143 |
+
# In[16]:
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
ov.utils.plot_paga(adata,basis='umap', size=50, alpha=.1,title='PAGA Slingshot-graph',
|
| 147 |
+
min_edge_width=2, node_size_scale=1.5,show=False,legend_loc=False)
|
| 148 |
+
|
| 149 |
+
|
| 150 |
+
# ## Trajectory inference with Palantir
|
| 151 |
+
#
|
| 152 |
+
# Palantir can be run by specifying an approxiate early cell.
|
| 153 |
+
#
|
| 154 |
+
# Palantir can automatically determine the terminal states as well. In this dataset, we know the terminal states and we will set them using the terminal_states parameter
|
| 155 |
+
#
|
| 156 |
+
# Here, we used `ov.single.TrajInfer` to construct a Trajectory Inference object.
|
| 157 |
+
|
| 158 |
+
# In[17]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
Traj=ov.single.TrajInfer(adata,basis='X_umap',groupby='clusters',
|
| 162 |
+
use_rep='scaled|original|X_pca',n_comps=50)
|
| 163 |
+
Traj.set_origin_cells('nIPC')
|
| 164 |
+
Traj.set_terminal_cells(["Granule mature","OL","Astrocytes"])
|
| 165 |
+
|
| 166 |
+
|
| 167 |
+
# In[18]:
|
| 168 |
+
|
| 169 |
+
|
| 170 |
+
Traj.inference(method='palantir',num_waypoints=500)
|
| 171 |
+
|
| 172 |
+
|
| 173 |
+
# Palantir results can be visualized on the tSNE or UMAP using the plot_palantir_results function
|
| 174 |
+
|
| 175 |
+
# In[19]:
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
Traj.palantir_plot_pseudotime(embedding_basis='X_umap',cmap='RdBu_r',s=3)
|
| 179 |
+
|
| 180 |
+
|
| 181 |
+
# Once the cells are selected, it's often helpful to visualize the selection on the pseudotime trajectory to ensure we've isolated the correct cells for our specific trend. We can do this using the plot_branch_selection function:
|
| 182 |
+
|
| 183 |
+
# In[20]:
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
Traj.palantir_cal_branch(eps=0)
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# In[22]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
ov.externel.palantir.plot.plot_trajectory(adata, "Granule mature",
|
| 193 |
+
cell_color="palantir_entropy",
|
| 194 |
+
n_arrows=10,
|
| 195 |
+
color="red",
|
| 196 |
+
scanpy_kwargs=dict(cmap="RdBu_r"),
|
| 197 |
+
)
|
| 198 |
+
|
| 199 |
+
|
| 200 |
+
# Palantir uses Mellon Function Estimator to determine the gene expression trends along different lineages. The marker trends can be determined using the following snippet. This computes the trends for all lineages. A subset of lineages can be used using the lineages parameter.
|
| 201 |
+
|
| 202 |
+
# In[23]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
gene_trends = Traj.palantir_cal_gene_trends(
|
| 206 |
+
layers="MAGIC_imputed_data",
|
| 207 |
+
)
|
| 208 |
+
|
| 209 |
+
|
| 210 |
+
# In[24]:
|
| 211 |
+
|
| 212 |
+
|
| 213 |
+
genes = ['Cdca3','Rasl10a','Mog','Aqp4']
|
| 214 |
+
Traj.palantir_plot_gene_trends(genes)
|
| 215 |
+
plt.show()
|
| 216 |
+
|
| 217 |
+
|
| 218 |
+
# We can also use paga to visualize the cell stages
|
| 219 |
+
|
| 220 |
+
# In[25]:
|
| 221 |
+
|
| 222 |
+
|
| 223 |
+
ov.utils.cal_paga(adata,use_time_prior='palantir_pseudotime',vkey='paga',
|
| 224 |
+
groups='clusters')
|
| 225 |
+
|
| 226 |
+
|
| 227 |
+
# In[26]:
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
ov.utils.plot_paga(adata,basis='umap', size=50, alpha=.1,title='PAGA LTNN-graph',
|
| 231 |
+
min_edge_width=2, node_size_scale=1.5,show=False,legend_loc=False)
|
| 232 |
+
|
| 233 |
+
|
| 234 |
+
# In[ ]:
|
| 235 |
+
|
| 236 |
+
|
| 237 |
+
|
| 238 |
+
|
ovrawm/t_via.txt
ADDED
|
@@ -0,0 +1,193 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Trajectory Inference with VIA
|
| 5 |
+
#
|
| 6 |
+
# VIA is a single-cell Trajectory Inference method that offers topology construction, pseudotimes, automated terminal state prediction and automated plotting of temporal gene dynamics along lineages. Here, we have improved the original author's colouring logic and user habits so that users can use the anndata object directly for analysis。
|
| 7 |
+
#
|
| 8 |
+
# We have completed this tutorial using the analysis provided by the original VIA authors.
|
| 9 |
+
#
|
| 10 |
+
# Paper: [Generalized and scalable trajectory inference in single-cell omics data with VIA](https://www.nature.com/articles/s41467-021-25773-3)
|
| 11 |
+
#
|
| 12 |
+
# Code: https://github.com/ShobiStassen/VIA
|
| 13 |
+
#
|
| 14 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1A2X23z_RLJaYLbXaiCbZa-fjNbuGACrD?usp=sharing
|
| 15 |
+
|
| 16 |
+
# In[1]:
|
| 17 |
+
|
| 18 |
+
|
| 19 |
+
import omicverse as ov
|
| 20 |
+
import scanpy as sc
|
| 21 |
+
import matplotlib.pyplot as plt
|
| 22 |
+
ov.utils.ov_plot_set()
|
| 23 |
+
|
| 24 |
+
|
| 25 |
+
# ## Data loading and preprocessing
|
| 26 |
+
#
|
| 27 |
+
# We have used the dataset scRNA_hematopoiesis provided by the authors for this analysis, noting that the data have been normalized and logarithmicized but not scaled.
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
adata = ov.single.scRNA_hematopoiesis()
|
| 33 |
+
sc.tl.pca(adata, svd_solver='arpack', n_comps=200)
|
| 34 |
+
adata
|
| 35 |
+
|
| 36 |
+
|
| 37 |
+
# ## Model construct and run
|
| 38 |
+
#
|
| 39 |
+
# We need to specify the cell feature vector `adata_key` used for VIA inference, which can be X_pca, X_scVI or X_glue, depending on the purpose of your analysis, here we use X_pca directly. We also need to specify how many components to be used, the components should not larger than the length of vector.
|
| 40 |
+
#
|
| 41 |
+
# Besides, we need to specify the `clusters` to be colored and calculate for VIA. If the `root_user` is None, it will be calculated the root cell automatically.
|
| 42 |
+
#
|
| 43 |
+
# We need to set `basis` argument stored in `adata.obsm`. An example setting `tsne` because it stored in `obsm: 'tsne', 'MAGIC_imputed_data', 'palantir_branch_probs', 'X_pca'`
|
| 44 |
+
#
|
| 45 |
+
# We also need to set `clusters` argument stored in `adata.obs`. It means the celltype key.
|
| 46 |
+
#
|
| 47 |
+
# Other explaination of argument and attributes could be found at https://pyvia.readthedocs.io/en/latest/Parameters%20and%20Attributes.html
|
| 48 |
+
|
| 49 |
+
# In[3]:
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
v0 = ov.single.pyVIA(adata=adata,adata_key='X_pca',adata_ncomps=80, basis='tsne',
|
| 53 |
+
clusters='label',knn=30,random_seed=4,root_user=[4823],)
|
| 54 |
+
|
| 55 |
+
v0.run()
|
| 56 |
+
|
| 57 |
+
|
| 58 |
+
# ## Visualize and analysis
|
| 59 |
+
#
|
| 60 |
+
# Before the subsequent analysis, we need to specify the colour of each cluster. Here we use sc.pl.embedding to automatically colour each cluster, if you need to specify your own colours, specify the palette parameter
|
| 61 |
+
|
| 62 |
+
# In[4]:
|
| 63 |
+
|
| 64 |
+
|
| 65 |
+
fig, ax = plt.subplots(1,1,figsize=(4,4))
|
| 66 |
+
sc.pl.embedding(
|
| 67 |
+
adata,
|
| 68 |
+
basis="tsne",
|
| 69 |
+
color=['label'],
|
| 70 |
+
frameon=False,
|
| 71 |
+
ncols=1,
|
| 72 |
+
wspace=0.5,
|
| 73 |
+
show=False,
|
| 74 |
+
ax=ax
|
| 75 |
+
)
|
| 76 |
+
fig.savefig('figures/via_fig1.png',dpi=300,bbox_inches = 'tight')
|
| 77 |
+
|
| 78 |
+
|
| 79 |
+
# ## VIA graph
|
| 80 |
+
#
|
| 81 |
+
# To visualize the results of the Trajectory inference in various ways. Via offers various plotting functions.We first show the cluster-graph level trajectory abstraction consisting of two subplots colored by annotated (true_label) composition and by pseudotime
|
| 82 |
+
|
| 83 |
+
# In[5]:
|
| 84 |
+
|
| 85 |
+
|
| 86 |
+
fig, ax, ax1 = v0.plot_piechart_graph(clusters='label',cmap='Reds',dpi=80,
|
| 87 |
+
show_legend=False,ax_text=False,fontsize=4)
|
| 88 |
+
fig.savefig('figures/via_fig2.png',dpi=300,bbox_inches = 'tight')
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# In[ ]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
#you can use `v0.model.single_cell_pt_markov` to extract the pseudotime
|
| 95 |
+
v0.get_pseudotime(v0.adata)
|
| 96 |
+
v0.adata
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
# ## Visualise gene/feature graph
|
| 100 |
+
#
|
| 101 |
+
# View the gene expression along the VIA graph. We use the computed HNSW small world graph in VIA to accelerate the gene imputation calculations (using similar approach to MAGIC) as follows:
|
| 102 |
+
#
|
| 103 |
+
|
| 104 |
+
# In[6]:
|
| 105 |
+
|
| 106 |
+
|
| 107 |
+
gene_list_magic = ['IL3RA', 'IRF8', 'GATA1', 'GATA2', 'ITGA2B', 'MPO', 'CD79B', 'SPI1', 'CD34', 'CSF1R', 'ITGAX']
|
| 108 |
+
fig,axs=v0.plot_clustergraph(gene_list=gene_list_magic[:4],figsize=(12,3),)
|
| 109 |
+
fig.savefig('figures/via_fig2_1.png',dpi=300,bbox_inches = 'tight')
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
# ## Trajectory projection
|
| 113 |
+
#
|
| 114 |
+
# Visualize the overall VIA trajectory projected onto a 2D embedding (UMAP, Phate, TSNE etc) in different ways.
|
| 115 |
+
#
|
| 116 |
+
# - Draw the high-level clustergraph abstraction onto the embedding;
|
| 117 |
+
# - Draws a vector field plot of the more fine-grained directionality of cells along the trajectory projected onto an embedding.
|
| 118 |
+
# - Draw high-edge resolution directed graph
|
| 119 |
+
|
| 120 |
+
# In[7]:
|
| 121 |
+
|
| 122 |
+
|
| 123 |
+
fig,ax1,ax2=v0.plot_trajectory_gams(basis='tsne',clusters='label',draw_all_curves=False)
|
| 124 |
+
fig.savefig('figures/via_fig3.png',dpi=300,bbox_inches = 'tight')
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
# In[8]:
|
| 128 |
+
|
| 129 |
+
|
| 130 |
+
fig,ax=v0.plot_stream(basis='tsne',clusters='label',
|
| 131 |
+
density_grid=0.8, scatter_size=30, scatter_alpha=0.3, linewidth=0.5)
|
| 132 |
+
fig.savefig('figures/via_fig4.png',dpi=300,bbox_inches = 'tight')
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
# In[9]:
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
fig,ax=v0.plot_stream(basis='tsne',density_grid=0.8, scatter_size=30, color_scheme='time', linewidth=0.5,
|
| 139 |
+
min_mass = 1, cutoff_perc = 5, scatter_alpha=0.3, marker_edgewidth=0.1,
|
| 140 |
+
density_stream = 2, smooth_transition=1, smooth_grid=0.5)
|
| 141 |
+
fig.savefig('figures/via_fig5.png',dpi=300,bbox_inches = 'tight')
|
| 142 |
+
|
| 143 |
+
|
| 144 |
+
# ## Probabilistic pathways
|
| 145 |
+
#
|
| 146 |
+
# Visualize the probabilistic pathways from root to terminal state as indicated by the lineage likelihood. The higher the linage likelihood, the greater the potential of that particular cell to differentiate towards the terminal state of interest.
|
| 147 |
+
|
| 148 |
+
# In[10]:
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
fig,axs=v0.plot_lineage_probability(figsize=(8,4),)
|
| 152 |
+
fig.savefig('figures/via_fig6.png',dpi=300,bbox_inches = 'tight')
|
| 153 |
+
|
| 154 |
+
|
| 155 |
+
# We can specify a specific linkage for visualisation
|
| 156 |
+
|
| 157 |
+
# In[11]:
|
| 158 |
+
|
| 159 |
+
|
| 160 |
+
fig,axs=v0.plot_lineage_probability(figsize=(6,3),marker_lineages = [2,3])
|
| 161 |
+
fig.savefig('figures/via_fig7.png',dpi=300,bbox_inches = 'tight')
|
| 162 |
+
|
| 163 |
+
|
| 164 |
+
# ## Gene Dynamics
|
| 165 |
+
#
|
| 166 |
+
# The gene dynamics along pseudotime for all detected lineages are automatically inferred by VIA. These can be interpreted as the change in gene expression along any given lineage.
|
| 167 |
+
|
| 168 |
+
# In[12]:
|
| 169 |
+
|
| 170 |
+
|
| 171 |
+
fig,axs=v0.plot_gene_trend(gene_list=gene_list_magic,figsize=(8,6),)
|
| 172 |
+
fig.savefig('figures/via_fig8.png',dpi=300,bbox_inches = 'tight')
|
| 173 |
+
|
| 174 |
+
|
| 175 |
+
# In[14]:
|
| 176 |
+
|
| 177 |
+
|
| 178 |
+
fig,ax=v0.plot_gene_trend_heatmap(gene_list=gene_list_magic,figsize=(4,4),
|
| 179 |
+
marker_lineages=[2])
|
| 180 |
+
fig.savefig('figures/via_fig9.png',dpi=300,bbox_inches = 'tight')
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
# In[ ]:
|
| 184 |
+
|
| 185 |
+
|
| 186 |
+
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
# In[ ]:
|
| 190 |
+
|
| 191 |
+
|
| 192 |
+
|
| 193 |
+
|
ovrawm/t_via_velo.txt
ADDED
|
@@ -0,0 +1,102 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Trajectory Inference with VIA and scVelo
|
| 5 |
+
#
|
| 6 |
+
# When scRNA-velocity is available, it can be used to guide the trajectory inference and automate initial state prediction. However, because RNA velocitycan be misguided by(Bergen 2021) boosts in expression, variable transcription rates and data capture scope limited to steady-state populations only, users might find it useful to adjust the level of influence the RNA-velocity data should exercise on the inferred TI.
|
| 7 |
+
#
|
| 8 |
+
# Paper: [Generalized and scalable trajectory inference in single-cell omics data with VIA](https://www.nature.com/articles/s41467-021-25773-3)
|
| 9 |
+
#
|
| 10 |
+
# Code: https://github.com/ShobiStassen/VIA
|
| 11 |
+
#
|
| 12 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1MtGr3e9uUb_BWOzKlcbOTiCYsZpljEyF?usp=sharing
|
| 13 |
+
|
| 14 |
+
# In[7]:
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
import omicverse as ov
|
| 18 |
+
import scanpy as sc
|
| 19 |
+
import scvelo as scv
|
| 20 |
+
import cellrank as cr
|
| 21 |
+
ov.utils.ov_plot_set()
|
| 22 |
+
|
| 23 |
+
|
| 24 |
+
# ## Data loading and preprocessing
|
| 25 |
+
#
|
| 26 |
+
# We use a familiar endocrine-genesis dataset (Bastidas-Ponce et al. (2019).) to demonstrate initial state prediction at the EP Ngn3 low cells and automatic captures of the 4 differentiated islets (alpha, beta, delta and epsilon). As mentioned, it us useful to control the level of influence of RNA-velocity relative to gene-gene distance and this is done using the velo_weight parameter.
|
| 27 |
+
|
| 28 |
+
# In[2]:
|
| 29 |
+
|
| 30 |
+
|
| 31 |
+
adata = cr.datasets.pancreas()
|
| 32 |
+
adata
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
# In[3]:
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
n_pcs = 30
|
| 39 |
+
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=5000)
|
| 40 |
+
sc.tl.pca(adata, n_comps = n_pcs)
|
| 41 |
+
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
|
| 42 |
+
scv.tl.velocity(adata, mode='stochastic') # good results acheived with mode = 'stochastic' too
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# ## Initialize and run VIA
|
| 46 |
+
#
|
| 47 |
+
#
|
| 48 |
+
|
| 49 |
+
# In[4]:
|
| 50 |
+
|
| 51 |
+
|
| 52 |
+
v0 = ov.single.pyVIA(adata=adata,adata_key='X_pca',adata_ncomps=n_pcs, basis='X_umap',
|
| 53 |
+
clusters='clusters',knn=20, root_user=None,
|
| 54 |
+
dataset='', random_seed=42,is_coarse=True, preserve_disconnected=True, pseudotime_threshold_TS=50,
|
| 55 |
+
piegraph_arrow_head_width=0.15,piegraph_edgeweight_scalingfactor=2.5,
|
| 56 |
+
velocity_matrix=adata.layers['velocity'],gene_matrix=adata.X.todense(),velo_weight=0.5,
|
| 57 |
+
edgebundle_pruning_twice=False, edgebundle_pruning=0.15, pca_loadings = adata.varm['PCs']
|
| 58 |
+
)
|
| 59 |
+
|
| 60 |
+
v0.run()
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
# In[ ]:
|
| 64 |
+
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
|
| 68 |
+
|
| 69 |
+
# In[5]:
|
| 70 |
+
|
| 71 |
+
|
| 72 |
+
fig, ax, ax1 = v0.plot_piechart_graph(clusters='clusters',cmap='Reds',dpi=80,
|
| 73 |
+
show_legend=False,ax_text=False,fontsize=4)
|
| 74 |
+
fig.set_size_inches(8,4)
|
| 75 |
+
|
| 76 |
+
|
| 77 |
+
# ## Visualize trajectory and cell progression
|
| 78 |
+
#
|
| 79 |
+
# Fine grained vector fields
|
| 80 |
+
|
| 81 |
+
# In[8]:
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
v0.plot_trajectory_gams(basis='X_umap',clusters='clusters',draw_all_curves=False)
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
# In[9]:
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
v0.plot_stream(basis='X_umap',clusters='clusters',
|
| 91 |
+
density_grid=0.8, scatter_size=30, scatter_alpha=0.3, linewidth=0.5)
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
# ## Draw lineage likelihoods
|
| 95 |
+
#
|
| 96 |
+
# These indicate potential pathways corresponding to the 4 islets (two types of Beta islets Lineage 5 and 12)
|
| 97 |
+
|
| 98 |
+
# In[10]:
|
| 99 |
+
|
| 100 |
+
|
| 101 |
+
v0.plot_lineage_probability()
|
| 102 |
+
|
ovrawm/t_visualize_bulk.txt
ADDED
|
@@ -0,0 +1,170 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Visualization of Bulk RNA-seq
|
| 5 |
+
#
|
| 6 |
+
# In this part, we will introduce the tutorial of special plot of `omicverse`.
|
| 7 |
+
|
| 8 |
+
# In[1]:
|
| 9 |
+
|
| 10 |
+
|
| 11 |
+
import omicverse as ov
|
| 12 |
+
import scanpy as sc
|
| 13 |
+
import matplotlib.pyplot as plt
|
| 14 |
+
ov.plot_set()
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
# ## Venn plot
|
| 18 |
+
#
|
| 19 |
+
# In transcriptome analyses, we often have to study differential genes that are common to different groups. Here, we provide `ov.pl.venn` to draw venn plots to visualise differential genes.
|
| 20 |
+
#
|
| 21 |
+
# **Function**: `ov.pl.venn`:
|
| 22 |
+
# - sets: Subgroups requiring venn plots, Dictionary format, keys no more than 4
|
| 23 |
+
# - palette: You can also re-specify the colour bar that needs to be drawn, just set `palette=['#FFFFFF','#000000']`, we have prepared `ov.pl.red_color`,`ov.pl.blue_color`,`ov.pl.green_color`,`ov.pl.orange_color`, by default.
|
| 24 |
+
# - fontsize: the fontsize and linewidth to visualize, fontsize will be multiplied by 2
|
| 25 |
+
|
| 26 |
+
# In[20]:
|
| 27 |
+
|
| 28 |
+
|
| 29 |
+
fig,ax=plt.subplots(figsize = (4,4))
|
| 30 |
+
#dict of sets
|
| 31 |
+
sets = {
|
| 32 |
+
'Set1:name': {1,2,3},
|
| 33 |
+
'Set2': {1,2,3,4},
|
| 34 |
+
'Set3': {3,4},
|
| 35 |
+
'Set4': {5,6}
|
| 36 |
+
}
|
| 37 |
+
#plot venn
|
| 38 |
+
ov.pl.venn(sets=sets,palette=ov.pl.sc_color,
|
| 39 |
+
fontsize=5.5,ax=ax,
|
| 40 |
+
)
|
| 41 |
+
|
| 42 |
+
|
| 43 |
+
#If we need to annotate genes, we can use plt.annotate for this purpose,
|
| 44 |
+
#we need to modify the text content, xy and xytext parameters.
|
| 45 |
+
plt.annotate('gene1,gene2', xy=(50,30), xytext=(0,-100),
|
| 46 |
+
ha='center', textcoords='offset points',
|
| 47 |
+
bbox=dict(boxstyle='round,pad=0.5', fc='gray', alpha=0.1),
|
| 48 |
+
arrowprops=dict(arrowstyle='->', color='gray'),size=12)
|
| 49 |
+
|
| 50 |
+
#Set the title
|
| 51 |
+
plt.title('Venn4',fontsize=13)
|
| 52 |
+
|
| 53 |
+
#save figure
|
| 54 |
+
fig.savefig("figures/bulk_venn4.png",dpi=300,bbox_inches = 'tight')
|
| 55 |
+
|
| 56 |
+
|
| 57 |
+
# In[9]:
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
fig,ax=plt.subplots(figsize = (4,4))
|
| 61 |
+
#dict of sets
|
| 62 |
+
sets = {
|
| 63 |
+
'Set1:name': {1,2,3},
|
| 64 |
+
'Set2': {1,2,3,4},
|
| 65 |
+
'Set3': {3,4},
|
| 66 |
+
}
|
| 67 |
+
|
| 68 |
+
ov.pl.venn(sets=sets,ax=ax,fontsize=5.5,
|
| 69 |
+
palette=ov.pl.red_color)
|
| 70 |
+
|
| 71 |
+
plt.title('Venn3',fontsize=13)
|
| 72 |
+
|
| 73 |
+
|
| 74 |
+
# ## Volcano plot
|
| 75 |
+
#
|
| 76 |
+
# For differentially expressed genes, we tend to visualise them only with volcano plots. Here, we present a method for mapping volcanoes using Python `ov.pl.volcano`.
|
| 77 |
+
#
|
| 78 |
+
# **Function**: `ov.pl.venn`:
|
| 79 |
+
#
|
| 80 |
+
# main argument
|
| 81 |
+
# - result: the DEGs result
|
| 82 |
+
# - pval_name: the names of the columns whose vertical coordinates need to be plotted, stored in result.columns. In Bulk RNA-seq experiments, we usually set this to qvalue.
|
| 83 |
+
# - fc_name: The names of the columns for which you need to plot the horizontal coordinates, stored in result.columns. In Bulk RNA-seq experiments, we typically set this to log2FC.
|
| 84 |
+
# - fc_max: We need to set the threshold for the difference foldchange
|
| 85 |
+
# - fc_min: We need to set the threshold for the difference foldchange
|
| 86 |
+
# - pval_threshold: We need to set the threshold for the qvalue
|
| 87 |
+
# - pval_max: We also need to set boundary values so that the data is not too large to affect the visualisation
|
| 88 |
+
# - FC_max: We also need to set boundary values so that the data is not too large to affect the visualisation
|
| 89 |
+
#
|
| 90 |
+
# plot argument
|
| 91 |
+
# - figsize: The size of the generated figure, by default (4,4).
|
| 92 |
+
# - title: The title of the plot, by default ''.
|
| 93 |
+
# - titlefont: A dictionary of font properties for the plot title, by default {'weight':'normal','size':14,}.
|
| 94 |
+
# - up_color: The color of the up-regulated genes in the plot, by default '#e25d5d'.
|
| 95 |
+
# - down_color: The color of the down-regulated genes in the plot, by default '#7388c1'.
|
| 96 |
+
# - normal_color: The color of the non-significant genes in the plot, by default '#d7d7d7'.
|
| 97 |
+
# - legend_bbox: A tuple containing the coordinates of the legend's bounding box, by default (0.8, -0.2).
|
| 98 |
+
# - legend_ncol: The number of columns in the legend, by default 2.
|
| 99 |
+
# - legend_fontsize: The font size of the legend, by default 12.
|
| 100 |
+
# - plot_genes: A list of genes to be plotted on the volcano plot, by default None.
|
| 101 |
+
# - plot_genes_num: The number of genes to be plotted on the volcano plot, by default 10.
|
| 102 |
+
# - plot_genes_fontsize: The font size of the genes to be plotted on the volcano plot, by default 10.
|
| 103 |
+
# - ticks_fontsize: The font size of the ticks, by default 12.
|
| 104 |
+
|
| 105 |
+
# In[3]:
|
| 106 |
+
|
| 107 |
+
|
| 108 |
+
result=ov.read('data/dds_result.csv',index_col=0)
|
| 109 |
+
result.head()
|
| 110 |
+
|
| 111 |
+
|
| 112 |
+
# In[4]:
|
| 113 |
+
|
| 114 |
+
|
| 115 |
+
ov.pl.volcano(result,pval_name='qvalue',fc_name='log2FoldChange',
|
| 116 |
+
pval_threshold=0.05,fc_max=1.5,fc_min=-1.5,
|
| 117 |
+
pval_max=10,FC_max=10,
|
| 118 |
+
figsize=(4,4),title='DEGs in Bulk',titlefont={'weight':'normal','size':14,},
|
| 119 |
+
up_color='#e25d5d',down_color='#7388c1',normal_color='#d7d7d7',
|
| 120 |
+
up_fontcolor='#e25d5d',down_fontcolor='#7388c1',normal_fontcolor='#d7d7d7',
|
| 121 |
+
legend_bbox=(0.8, -0.2),legend_ncol=2,legend_fontsize=12,
|
| 122 |
+
plot_genes=None,plot_genes_num=10,plot_genes_fontsize=11,
|
| 123 |
+
ticks_fontsize=12,)
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
# ## Box plot
|
| 127 |
+
#
|
| 128 |
+
# For differentially expressed genes in different groups, we sometimes need to compare the differences between different groups, and this is when we need to use box-and-line plots to do the comparison
|
| 129 |
+
#
|
| 130 |
+
# **Function**: `ov.pl.boxplot`:
|
| 131 |
+
#
|
| 132 |
+
# - data: the data to visualize the boxplt example could be found in `seaborn.load_dataset("tips")`
|
| 133 |
+
# - x_value, y_value, hue: Inputs for plotting long-form data. See examples for interpretation.
|
| 134 |
+
# - figsize: The size of the generated figure, by default (4,4).
|
| 135 |
+
# - fontsize: The font size of the tick and labels, by default 12.
|
| 136 |
+
# - title: The title of the plot, by default ''.
|
| 137 |
+
#
|
| 138 |
+
#
|
| 139 |
+
# **Function**: `ov.pl.add_palue`:
|
| 140 |
+
# - ax: the axes of bardotplot
|
| 141 |
+
# - line_x1: The left side of the p-value line to be plotted
|
| 142 |
+
# - line_x2: The right side of the p-value line to be plotted|
|
| 143 |
+
# - line_y: The height of the p-value line to be plotted
|
| 144 |
+
# - text_y: How much above the p-value line is plotted text
|
| 145 |
+
# - text: the text of p-value, you can set `***` to instead `p<0.001`
|
| 146 |
+
# - fontsize: the fontsize of text
|
| 147 |
+
# - fontcolor: the color of text
|
| 148 |
+
# - horizontalalignment: the location of text
|
| 149 |
+
|
| 150 |
+
# In[3]:
|
| 151 |
+
|
| 152 |
+
|
| 153 |
+
import seaborn as sns
|
| 154 |
+
data = sns.load_dataset("tips")
|
| 155 |
+
data.head()
|
| 156 |
+
|
| 157 |
+
|
| 158 |
+
# In[7]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
fig,ax=ov.pl.boxplot(data,hue='sex',x_value='day',y_value='total_bill',
|
| 162 |
+
palette=ov.pl.red_color,
|
| 163 |
+
figsize=(4,2),fontsize=12,title='Tips',)
|
| 164 |
+
|
| 165 |
+
ov.pl.add_palue(ax,line_x1=-0.5,line_x2=0.5,line_y=40,
|
| 166 |
+
text_y=0.2,
|
| 167 |
+
text='$p={}$'.format(round(0.001,3)),
|
| 168 |
+
fontsize=11,fontcolor='#000000',
|
| 169 |
+
horizontalalignment='center',)
|
| 170 |
+
|
ovrawm/t_visualize_colorsystem.txt
ADDED
|
@@ -0,0 +1,223 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Color system
|
| 5 |
+
#
|
| 6 |
+
# In OmicVerse, we offer a color system based on Eastern aesthetics, featuring 384 representative colors derived from the Forbidden City. We will utilize these colors in combination for future visualizations.
|
| 7 |
+
#
|
| 8 |
+
# All color come from the book: "中国传统色:故宫里的色彩美学" (ISBN: 9787521716054)
|
| 9 |
+
|
| 10 |
+
# In[ ]:
|
| 11 |
+
|
| 12 |
+
|
| 13 |
+
import omicverse as ov
|
| 14 |
+
import scanpy as sc
|
| 15 |
+
#import scvelo as scv
|
| 16 |
+
ov.plot_set()
|
| 17 |
+
|
| 18 |
+
|
| 19 |
+
# We utilized single-cell RNA-seq data (GEO accession: GSE95753) obtained from the dentate gyrus of the hippocampus in mouse.
|
| 20 |
+
|
| 21 |
+
# In[2]:
|
| 22 |
+
|
| 23 |
+
|
| 24 |
+
adata = ov.read('data/DentateGyrus/10X43_1.h5ad')
|
| 25 |
+
adata
|
| 26 |
+
|
| 27 |
+
|
| 28 |
+
# ## Understanding the Color System
|
| 29 |
+
#
|
| 30 |
+
# In OmicVerse, we offer a color system based on Eastern aesthetics, featuring 384 representative colors derived from the Forbidden City. We will utilize these colors in combination for future visualizations.
|
| 31 |
+
|
| 32 |
+
# In[3]:
|
| 33 |
+
|
| 34 |
+
|
| 35 |
+
fb=ov.pl.ForbiddenCity()
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
# In[7]:
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
from IPython.display import HTML
|
| 42 |
+
HTML(fb.visual_color(loc_range=(0,384),
|
| 43 |
+
num_per_row=24))
|
| 44 |
+
|
| 45 |
+
|
| 46 |
+
# we can get a color using `get_color`
|
| 47 |
+
|
| 48 |
+
# In[14]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
fb.get_color(name='凝夜紫')
|
| 52 |
+
|
| 53 |
+
|
| 54 |
+
# ## Default Colormap
|
| 55 |
+
#
|
| 56 |
+
# We have provided a range of default colors including `green`, `red`, `pink`, `purple`, `yellow`, `brown`, `blue`, and `grey`. Each of these colors comes with its own set of sub-colormaps, providing a more granular level of color differentiation.
|
| 57 |
+
#
|
| 58 |
+
# Here's a breakdown of the sub-colormaps available for each default color:
|
| 59 |
+
#
|
| 60 |
+
# - **Green**:
|
| 61 |
+
# - `green1`: `Forbidden_Cmap(range(1, 19))`
|
| 62 |
+
# - `green2`: `Forbidden_Cmap(range(19, 41))`
|
| 63 |
+
# - `green3`: `Forbidden_Cmap(range(41, 62))`
|
| 64 |
+
#
|
| 65 |
+
# - **Red**:
|
| 66 |
+
# - `red1`: `Forbidden_Cmap(range(62, 77))`
|
| 67 |
+
# - `red2`: `Forbidden_Cmap(range(77, 104))`
|
| 68 |
+
#
|
| 69 |
+
# - **Pink**:
|
| 70 |
+
# - `pink1`: `Forbidden_Cmap(range(104, 134))`
|
| 71 |
+
# - `pink2`: `Forbidden_Cmap(range(134, 148))`
|
| 72 |
+
#
|
| 73 |
+
# - **Purple**:
|
| 74 |
+
# - `purple1`: `Forbidden_Cmap(range(148, 162))`
|
| 75 |
+
# - `purple2`: `Forbidden_Cmap(range(162, 176))`
|
| 76 |
+
#
|
| 77 |
+
# - **Yellow**:
|
| 78 |
+
# - `yellow1`: `Forbidden_Cmap(range(176, 196))`
|
| 79 |
+
# - `yellow2`: `Forbidden_Cmap(range(196, 207))`
|
| 80 |
+
# - `yellow3`: `Forbidden_Cmap(range(255, 276))`
|
| 81 |
+
#
|
| 82 |
+
# - **Brown**:
|
| 83 |
+
# - `brown1`: `Forbidden_Cmap(range(207, 228))`
|
| 84 |
+
# - `brown2`: `Forbidden_Cmap(range(228, 246))`
|
| 85 |
+
# - `brown3`: `Forbidden_Cmap(range(246, 255))`
|
| 86 |
+
# - `brown4`: `Forbidden_Cmap(range(276, 293))`
|
| 87 |
+
#
|
| 88 |
+
# - **Blue**:
|
| 89 |
+
# - `blue1`: `Forbidden_Cmap(range(293, 312))`
|
| 90 |
+
# - `blue2`: `Forbidden_Cmap(range(312, 321))`
|
| 91 |
+
# - `blue3`: `Forbidden_Cmap(range(321, 333))`
|
| 92 |
+
# - `blue4`: `Forbidden_Cmap(range(333, 339))`
|
| 93 |
+
#
|
| 94 |
+
# - **Grey**:
|
| 95 |
+
# - `grey1`: `Forbidden_Cmap(range(339, 356))`
|
| 96 |
+
# - `grey2`: `Forbidden_Cmap(range(356, 385))`
|
| 97 |
+
#
|
| 98 |
+
# Each main color can be represented as a combination of its sub-colormaps:
|
| 99 |
+
#
|
| 100 |
+
# - `green = green1 + green2 + green3`
|
| 101 |
+
# - `red = red1 + red2`
|
| 102 |
+
# - `pink = pink1 + pink2`
|
| 103 |
+
# - `purple = purple1 + purple2`
|
| 104 |
+
# - `yellow = yellow1 + yellow2 + yellow3`
|
| 105 |
+
# - `brown = brown1 + brown2 + brown3 + brown4`
|
| 106 |
+
# - `blue = blue1 + blue2 + blue3 + blue4`
|
| 107 |
+
# - `grey = grey1 + grey2`
|
| 108 |
+
#
|
| 109 |
+
# These colormaps can be utilized in various applications where color differentiation is necessary, providing flexibility in visual representation.
|
| 110 |
+
#
|
| 111 |
+
# `palette` is the argument we need to revise
|
| 112 |
+
|
| 113 |
+
# In[13]:
|
| 114 |
+
|
| 115 |
+
|
| 116 |
+
import matplotlib.pyplot as plt
|
| 117 |
+
fig, axes = plt.subplots(1,3,figsize=(9,3))
|
| 118 |
+
ov.pl.embedding(adata,
|
| 119 |
+
basis='X_umap',
|
| 120 |
+
frameon='small',
|
| 121 |
+
color=["clusters"],
|
| 122 |
+
palette=fb.red[:],
|
| 123 |
+
ncols=3,
|
| 124 |
+
show=False,
|
| 125 |
+
legend_loc=None,
|
| 126 |
+
ax=axes[0])
|
| 127 |
+
|
| 128 |
+
ov.pl.embedding(adata,
|
| 129 |
+
basis='X_umap',
|
| 130 |
+
frameon='small',
|
| 131 |
+
color=["clusters"],
|
| 132 |
+
palette=fb.pink1[:],
|
| 133 |
+
ncols=3,show=False,
|
| 134 |
+
legend_loc=None,
|
| 135 |
+
ax=axes[1])
|
| 136 |
+
|
| 137 |
+
ov.pl.embedding(adata,
|
| 138 |
+
basis='X_umap',
|
| 139 |
+
frameon='small',
|
| 140 |
+
color=["clusters"],
|
| 141 |
+
palette=fb.red1[:4]+fb.blue1,
|
| 142 |
+
ncols=3,show=False,
|
| 143 |
+
ax=axes[2])
|
| 144 |
+
|
| 145 |
+
|
| 146 |
+
|
| 147 |
+
|
| 148 |
+
# In[31]:
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
color_dict={'Astrocytes': '#e40414',
|
| 152 |
+
'Cajal Retzius': '#ec5414',
|
| 153 |
+
'Cck-Tox': '#ec4c2c',
|
| 154 |
+
'Endothelial': '#d42c24',
|
| 155 |
+
'GABA': '#2c5ca4',
|
| 156 |
+
'Granule immature': '#acd4ec',
|
| 157 |
+
'Granule mature': '#a4bcdc',
|
| 158 |
+
'Microglia': '#8caccc',
|
| 159 |
+
'Mossy': '#8cacdc',
|
| 160 |
+
'Neuroblast': '#6c9cc4',
|
| 161 |
+
'OL': '#6c94cc',
|
| 162 |
+
'OPC': '#5c74bc',
|
| 163 |
+
'Radial Glia-like': '#4c94c4',
|
| 164 |
+
'nIPC': '#3474ac'}
|
| 165 |
+
|
| 166 |
+
ov.pl.embedding(adata,
|
| 167 |
+
basis='X_umap',
|
| 168 |
+
frameon='small',
|
| 169 |
+
color=["clusters"],
|
| 170 |
+
palette=color_dict,
|
| 171 |
+
ncols=3,show=False,
|
| 172 |
+
)
|
| 173 |
+
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
# ## Segmented Colormap
|
| 177 |
+
#
|
| 178 |
+
# When we need to create a continuous color gradient, we will use another function: `get_cmap_seg`, and we can combine the colors we need for visualization.
|
| 179 |
+
|
| 180 |
+
# In[22]:
|
| 181 |
+
|
| 182 |
+
|
| 183 |
+
colors=[
|
| 184 |
+
fb.get_color_rgb('群青'),
|
| 185 |
+
fb.get_color_rgb('半见'),
|
| 186 |
+
fb.get_color_rgb('丹罽'),
|
| 187 |
+
]
|
| 188 |
+
fb.get_cmap_seg(colors)
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
# In[24]:
|
| 192 |
+
|
| 193 |
+
|
| 194 |
+
colors=[
|
| 195 |
+
fb.get_color_rgb('群青'),
|
| 196 |
+
fb.get_color_rgb('山矾'),
|
| 197 |
+
fb.get_color_rgb('丹罽'),
|
| 198 |
+
]
|
| 199 |
+
fb.get_cmap_seg(colors)
|
| 200 |
+
|
| 201 |
+
|
| 202 |
+
# In[25]:
|
| 203 |
+
|
| 204 |
+
|
| 205 |
+
colors=[
|
| 206 |
+
fb.get_color_rgb('山矾'),
|
| 207 |
+
fb.get_color_rgb('丹罽'),
|
| 208 |
+
]
|
| 209 |
+
fb.get_cmap_seg(colors)
|
| 210 |
+
|
| 211 |
+
|
| 212 |
+
# In[27]:
|
| 213 |
+
|
| 214 |
+
|
| 215 |
+
ov.pl.embedding(adata,
|
| 216 |
+
basis='X_umap',
|
| 217 |
+
frameon='small',
|
| 218 |
+
color=["Sox7"],
|
| 219 |
+
cmap=fb.get_cmap_seg(colors),
|
| 220 |
+
ncols=3,show=False,
|
| 221 |
+
#vmin=-1,vmax=1
|
| 222 |
+
)
|
| 223 |
+
|
ovrawm/t_visualize_single.txt
ADDED
|
@@ -0,0 +1,534 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # Visualization of single cell RNA-seq
|
| 5 |
+
#
|
| 6 |
+
# In this part, we will introduce the tutorial of special plot of `omicverse`.
|
| 7 |
+
|
| 8 |
+
# In[1]:
|
| 9 |
+
|
| 10 |
+
|
| 11 |
+
import omicverse as ov
|
| 12 |
+
import scanpy as sc
|
| 13 |
+
#import scvelo as scv
|
| 14 |
+
ov.plot_set()
|
| 15 |
+
|
| 16 |
+
|
| 17 |
+
# We utilized single-cell RNA-seq data (GEO accession: GSE95753) obtained from the dentate gyrus of the hippocampus in mouse.
|
| 18 |
+
|
| 19 |
+
# In[2]:
|
| 20 |
+
|
| 21 |
+
|
| 22 |
+
adata = ov.read('data/DentateGyrus/10X43_1.h5ad')
|
| 23 |
+
adata
|
| 24 |
+
|
| 25 |
+
|
| 26 |
+
# ## Optimizing color mapping
|
| 27 |
+
#
|
| 28 |
+
# Visualizing spatially resolved biological data with appropriate color mapping can significantly facilitate the exploration of underlying patterns and heterogeneity. Spaco (spatial colorization) provides a spatially constrained approach that generates discriminate color assignments for visualizing single-cell spatial data in various scenarios.
|
| 29 |
+
#
|
| 30 |
+
# Jing Z, Zhu Q, Li L, Xie Y, Wu X, Fang Q, et al. [Spaco: A comprehensive tool for coloring spatial data at single-cell resolution.](https://doi.org/10.1016/j.patter.2023.100915) Patterns. 2024;100915
|
| 31 |
+
#
|
| 32 |
+
#
|
| 33 |
+
# **Function**: `ov.pl.optim_palette`:
|
| 34 |
+
# - adata: the datasets of scRNA-seq
|
| 35 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 36 |
+
# - colors: Specify the colour to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 37 |
+
# - palette: You can also re-specify the colour bar that needs to be drawn, just set `palette=['#FFFFFF','#000000']`, we have prepared `ov.pl.red_color`,`ov.pl.blue_color`,`ov.pl.green_color`,`ov.pl.orange_color`, by default.
|
| 38 |
+
|
| 39 |
+
# In[ ]:
|
| 40 |
+
|
| 41 |
+
|
| 42 |
+
optim_palette=ov.pl.optim_palette(adata,basis='X_umap',colors='clusters')
|
| 43 |
+
|
| 44 |
+
|
| 45 |
+
# In[4]:
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
import matplotlib.pyplot as plt
|
| 49 |
+
fig,ax=plt.subplots(figsize = (4,4))
|
| 50 |
+
ov.pl.embedding(adata,
|
| 51 |
+
basis='X_umap',
|
| 52 |
+
color='clusters',
|
| 53 |
+
frameon='small',
|
| 54 |
+
show=False,
|
| 55 |
+
palette=optim_palette,
|
| 56 |
+
ax=ax,)
|
| 57 |
+
plt.title('Cell Type of DentateGyrus',fontsize=15)
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
# In[5]:
|
| 61 |
+
|
| 62 |
+
|
| 63 |
+
ov.pl.embedding(adata,
|
| 64 |
+
basis='X_umap',
|
| 65 |
+
color='age(days)',
|
| 66 |
+
frameon='small',
|
| 67 |
+
show=False,)
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
# ## Stacked histogram of cell proportions
|
| 71 |
+
#
|
| 72 |
+
# This is a graph that appears widely in various CNS-level journals, and is limited to the fact that `scanpy` does not have a proper way of plotting it, and we provide `ov.pl.cellproportion` for plotting it here.
|
| 73 |
+
#
|
| 74 |
+
# **Function**: `ov.pl.cellproportion`:
|
| 75 |
+
# - adata: the datasets of scRNA-seq
|
| 76 |
+
# - celltype_clusters: Specify the colour to plot, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 77 |
+
# - groupby: The group variable for the different groups of cell types we need to display, in this case we are displaying different ages, so we set it to `age(days)`
|
| 78 |
+
# - groupby_li: If there are too many groups, we can also select the ones we are interested in plotting, here we use groupby_li to plot the groups
|
| 79 |
+
# - figsize: If we specify axes, then this variable can be left empty
|
| 80 |
+
# - legend: Whether to show a legend
|
| 81 |
+
|
| 82 |
+
# In[6]:
|
| 83 |
+
|
| 84 |
+
|
| 85 |
+
import matplotlib.pyplot as plt
|
| 86 |
+
fig,ax=plt.subplots(figsize = (1,4))
|
| 87 |
+
ov.pl.cellproportion(adata=adata,celltype_clusters='clusters',
|
| 88 |
+
groupby='age(days)',legend=True,ax=ax)
|
| 89 |
+
|
| 90 |
+
|
| 91 |
+
# In[7]:
|
| 92 |
+
|
| 93 |
+
|
| 94 |
+
fig,ax=plt.subplots(figsize = (2,2))
|
| 95 |
+
ov.pl.cellproportion(adata=adata,celltype_clusters='age(days)',
|
| 96 |
+
groupby='clusters',groupby_li=['nIPC','Granule immature','Granule mature'],
|
| 97 |
+
legend=True,ax=ax)
|
| 98 |
+
|
| 99 |
+
|
| 100 |
+
# If you are interested in the changes in cell types in different groups, we recommend using a stacked area graph.
|
| 101 |
+
|
| 102 |
+
# In[8]:
|
| 103 |
+
|
| 104 |
+
|
| 105 |
+
fig,ax=plt.subplots(figsize = (2,2))
|
| 106 |
+
ov.pl.cellstackarea(adata=adata,celltype_clusters='age(days)',
|
| 107 |
+
groupby='clusters',groupby_li=['nIPC','Granule immature','Granule mature'],
|
| 108 |
+
legend=True,ax=ax)
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
# ## A collection of some interesting embedded plot
|
| 112 |
+
#
|
| 113 |
+
# Our first presentation is an embedding map with the number and proportion of cell types. This graph visualises the low-dimensional representation of cells in addition to the number of cell proportions, etc. It should be noted that the cell proportions plotted on the left side may be distorted when there are too many cell types, and we would be grateful if anyone would be interested in fixing this bug.
|
| 114 |
+
#
|
| 115 |
+
# **Function**: `ov.pl.embedding_celltype`:
|
| 116 |
+
# - adata: the datasets of scRNA-seq
|
| 117 |
+
# - figsize: Note that we don't usually provide the ax parameter for combinatorial graphs, this is due to the fact that combinatorial graphs are made up of multiple axes, so the figsize parameter is more important, here we set it to `figsize=(7,4)`.
|
| 118 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 119 |
+
# - celltype_key: Specify the colour to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 120 |
+
# - title: Note that the space entered in title is used to control the position.
|
| 121 |
+
# - celltype_range: Since our number of cell types is different in each data, we want to have the flexibility to control where the cell scale plot is drawn, here we set it to `(1,10)`. You can also tweak the observations yourself to find the parameter that best suits your data.
|
| 122 |
+
# - embedding_range: As with the positional parameters of the cell types, they need to be adjusted several times on their own for optimal results.
|
| 123 |
+
|
| 124 |
+
# In[9]:
|
| 125 |
+
|
| 126 |
+
|
| 127 |
+
ov.pl.embedding_celltype(adata,figsize=(7,4),basis='X_umap',
|
| 128 |
+
celltype_key='clusters',
|
| 129 |
+
title=' Cell type',
|
| 130 |
+
celltype_range=(1,10),
|
| 131 |
+
embedding_range=(4,10),)
|
| 132 |
+
|
| 133 |
+
|
| 134 |
+
# Sometimes we want to be able to circle a certain type of cell that we are interested in, and here we use convex polygons to achieve this, while the shape of the convex polygons may be optimised in future versions.
|
| 135 |
+
#
|
| 136 |
+
# **Function**: `ov.pl.ConvexHull`:
|
| 137 |
+
# - adata: the datasets of scRNA-seq
|
| 138 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 139 |
+
# - cluster_key: Specify the celltype to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 140 |
+
# - hull_cluster: the target celltype to be circled.
|
| 141 |
+
|
| 142 |
+
# In[10]:
|
| 143 |
+
|
| 144 |
+
|
| 145 |
+
import matplotlib.pyplot as plt
|
| 146 |
+
fig,ax=plt.subplots(figsize = (4,4))
|
| 147 |
+
|
| 148 |
+
ov.pl.embedding(adata,
|
| 149 |
+
basis='X_umap',
|
| 150 |
+
color=['clusters'],
|
| 151 |
+
frameon='small',
|
| 152 |
+
show=False,
|
| 153 |
+
ax=ax)
|
| 154 |
+
|
| 155 |
+
ov.pl.ConvexHull(adata,
|
| 156 |
+
basis='X_umap',
|
| 157 |
+
cluster_key='clusters',
|
| 158 |
+
hull_cluster='Granule mature',
|
| 159 |
+
ax=ax)
|
| 160 |
+
|
| 161 |
+
|
| 162 |
+
# Besides, if you don't want to plot convexhull, you can plot the contour instead.
|
| 163 |
+
#
|
| 164 |
+
# **Function**: `ov.pl.contour`:
|
| 165 |
+
# - adata: the datasets of scRNA-seq
|
| 166 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 167 |
+
# - groupby: Specify the celltype to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 168 |
+
# - clusters: the target celltype to be circled.
|
| 169 |
+
# - colors: the color of the contour
|
| 170 |
+
# - linestyles: the linestyles of the contour
|
| 171 |
+
# - **kwargs: more kwargs could be found from `plt.contour`
|
| 172 |
+
|
| 173 |
+
# In[11]:
|
| 174 |
+
|
| 175 |
+
|
| 176 |
+
import matplotlib.pyplot as plt
|
| 177 |
+
fig,ax=plt.subplots(figsize = (4,4))
|
| 178 |
+
|
| 179 |
+
ov.pl.embedding(adata,
|
| 180 |
+
basis='X_umap',
|
| 181 |
+
color=['clusters'],
|
| 182 |
+
frameon='small',
|
| 183 |
+
show=False,
|
| 184 |
+
ax=ax)
|
| 185 |
+
|
| 186 |
+
ov.pl.contour(ax=ax,adata=adata,groupby='clusters',clusters=['Granule immature','Granule mature'],
|
| 187 |
+
basis='X_umap',contour_threshold=0.1,colors='#000000',
|
| 188 |
+
linestyles='dashed',)
|
| 189 |
+
|
| 190 |
+
|
| 191 |
+
# In scanpy's default `embedding` plotting function, when we set legend=True, legend masking may occur. To solve this problem, we introduced `ov.pl.embedding_adjust` in omicverse to automatically adjust the position of the legend.
|
| 192 |
+
#
|
| 193 |
+
# **Function**: `ov.pl.embedding_adjust`:
|
| 194 |
+
# - adata: the datasets of scRNA-seq
|
| 195 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 196 |
+
# - groupby: Specify the celltype to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 197 |
+
# - exclude: We can specify which cell types are not to be plotted, in this case we set it to `OL`
|
| 198 |
+
# - adjust_kwargs: We can manually specify the parameters of [adjustText](https://adjusttext.readthedocs.io/en/latest/), the specific parameters see the documentation of adjustText, it should be noted that we have to use dict to specify the parameters here.
|
| 199 |
+
# - text_kwargs: We can also specify the font colour manually by specifying the [text_kwargs](https://matplotlib.org/stable/api/_as_gen/matplotlib.pyplot.text.html) parameter
|
| 200 |
+
|
| 201 |
+
# In[12]:
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
from matplotlib import patheffects
|
| 205 |
+
import matplotlib.pyplot as plt
|
| 206 |
+
fig, ax = plt.subplots(figsize=(4,4))
|
| 207 |
+
|
| 208 |
+
ov.pl.embedding(adata,
|
| 209 |
+
basis='X_umap',
|
| 210 |
+
color=['clusters'],
|
| 211 |
+
show=False, legend_loc=None, add_outline=False,
|
| 212 |
+
frameon='small',legend_fontoutline=2,ax=ax
|
| 213 |
+
)
|
| 214 |
+
|
| 215 |
+
ov.pl.embedding_adjust(
|
| 216 |
+
adata,
|
| 217 |
+
groupby='clusters',
|
| 218 |
+
exclude=("OL",),
|
| 219 |
+
basis='X_umap',
|
| 220 |
+
ax=ax,
|
| 221 |
+
adjust_kwargs=dict(arrowprops=dict(arrowstyle='-', color='black')),
|
| 222 |
+
text_kwargs=dict(fontsize=12 ,weight='bold',
|
| 223 |
+
path_effects=[patheffects.withStroke(linewidth=2, foreground='w')] ),
|
| 224 |
+
)
|
| 225 |
+
|
| 226 |
+
|
| 227 |
+
# Sometimes we are interested in the distribution density of a certain class of cell types in a categorical variable, which is cumbersome to plot in the `scanpy` implementation, so we have simplified the implementation in omicverse and ensured the same plotting.
|
| 228 |
+
#
|
| 229 |
+
# **Function**: `ov.pl.embedding_density`:
|
| 230 |
+
# - adata: the datasets of scRNA-seq
|
| 231 |
+
# - basis: he position on the plane should be set to `X_spatial` in spatial RNA-seq, `X_umap`,`X_tsne`,`X_mde` in scRNA-seq and should not be set to `X_pca`
|
| 232 |
+
# - groupby: Specify the celltype to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 233 |
+
# - target_clusters: We can specify which cell types are to be plotted, in this case we set it to `Granule mature`
|
| 234 |
+
# - kwargs: other parameter can be found in `scanpy.pl.embedding`
|
| 235 |
+
|
| 236 |
+
# In[13]:
|
| 237 |
+
|
| 238 |
+
|
| 239 |
+
ov.pl.embedding_density(adata,
|
| 240 |
+
basis='X_umap',
|
| 241 |
+
groupby='clusters',
|
| 242 |
+
target_clusters='Granule mature',
|
| 243 |
+
frameon='small',
|
| 244 |
+
show=False,cmap='RdBu_r',alpha=0.8)
|
| 245 |
+
|
| 246 |
+
|
| 247 |
+
# ## Bar graph with overlapping dots (Bar-dot) plot
|
| 248 |
+
#
|
| 249 |
+
# In biological research, bardotplot plots are the most common class of graphs we use, but unfortunately, there is no direct implementation of plotting functions in either matplotlib, seaborn or scanpy. To compensate for this, we implement bardotplot plotting in omicverse and provide manual addition of p-values (it should be noted that manual addition refers to the manual addition of p-values for model fitting rather than making up p-values yourself).
|
| 250 |
+
|
| 251 |
+
# In[14]:
|
| 252 |
+
|
| 253 |
+
|
| 254 |
+
ov.single.geneset_aucell(adata,
|
| 255 |
+
geneset_name='Sox',
|
| 256 |
+
geneset=['Sox17', 'Sox4', 'Sox7', 'Sox18', 'Sox5'])
|
| 257 |
+
|
| 258 |
+
|
| 259 |
+
# In[15]:
|
| 260 |
+
|
| 261 |
+
|
| 262 |
+
ov.pl.embedding(adata,
|
| 263 |
+
basis='X_umap',
|
| 264 |
+
color=['Sox4'],
|
| 265 |
+
frameon='small',
|
| 266 |
+
show=False,)
|
| 267 |
+
|
| 268 |
+
|
| 269 |
+
# In[18]:
|
| 270 |
+
|
| 271 |
+
|
| 272 |
+
ov.pl.violin(adata,keys='Sox4',groupby='clusters',figsize=(6,3))
|
| 273 |
+
|
| 274 |
+
|
| 275 |
+
# **Function**: `ov.pl.embedding_density`:
|
| 276 |
+
# - adata: the datasets of scRNA-seq
|
| 277 |
+
# - groupby: Specify the celltype to be optimised, which should be for one of the columns in adata.obs, noting that it should have the colour first, and that we can use ov.pl.embedding to colour the cell types. If there is no colour then colour blind optimisation colour will be used.
|
| 278 |
+
# - color: The size of the variable to be plotted, which can be a gene, stored in adata.var, or a cell value, stored in adata.obs.
|
| 279 |
+
# - bar_kwargs: We provide the parameters of the barplot for input, see the matplotlib documentation for more [details](https://matplotlib.org/stable/api/_as_gen/matplotlib.pyplot.bar.html)
|
| 280 |
+
# - scatter_kwargs: We also provide the parameters of the scatter for input, see the matplotlib documentation for more [details](https://matplotlib.org/stable/api/_as_gen/matplotlib.pyplot.scatter.html)
|
| 281 |
+
#
|
| 282 |
+
# **Function**: `ov.pl.add_palue`:
|
| 283 |
+
# - ax: the axes of bardotplot
|
| 284 |
+
# - line_x1: The left side of the p-value line to be plotted
|
| 285 |
+
# - line_x2: The right side of the p-value line to be plotted|
|
| 286 |
+
# - line_y: The height of the p-value line to be plotted
|
| 287 |
+
# - text_y: How much above the p-value line is plotted text
|
| 288 |
+
# - text: the text of p-value, you can set `***` to instead `p<0.001`
|
| 289 |
+
# - fontsize: the fontsize of text
|
| 290 |
+
# - fontcolor: the color of text
|
| 291 |
+
# - horizontalalignment: the location of text
|
| 292 |
+
|
| 293 |
+
# In[19]:
|
| 294 |
+
|
| 295 |
+
|
| 296 |
+
fig, ax = plt.subplots(figsize=(6,2))
|
| 297 |
+
ov.pl.bardotplot(adata,groupby='clusters',color='Sox_aucell',figsize=(6,2),
|
| 298 |
+
ax=ax,
|
| 299 |
+
ylabel='Expression',
|
| 300 |
+
bar_kwargs={'alpha':0.5,'linewidth':2,'width':0.6,'capsize':4},
|
| 301 |
+
scatter_kwargs={'alpha':0.8,'s':10,'marker':'o'})
|
| 302 |
+
|
| 303 |
+
ov.pl.add_palue(ax,line_x1=3,line_x2=4,line_y=0.1,
|
| 304 |
+
text_y=0.02,
|
| 305 |
+
text='$p={}$'.format(round(0.001,3)),
|
| 306 |
+
fontsize=11,fontcolor='#000000',
|
| 307 |
+
horizontalalignment='center',)
|
| 308 |
+
|
| 309 |
+
|
| 310 |
+
# In[20]:
|
| 311 |
+
|
| 312 |
+
|
| 313 |
+
fig, ax = plt.subplots(figsize=(6,2))
|
| 314 |
+
ov.pl.bardotplot(adata,groupby='clusters',color='Sox17',figsize=(6,2),
|
| 315 |
+
ax=ax,
|
| 316 |
+
ylabel='Expression',xlabel='Cell Type',
|
| 317 |
+
bar_kwargs={'alpha':0.5,'linewidth':2,'width':0.6,'capsize':4},
|
| 318 |
+
scatter_kwargs={'alpha':0.8,'s':10,'marker':'o'})
|
| 319 |
+
|
| 320 |
+
ov.pl.add_palue(ax,line_x1=3,line_x2=4,line_y=2,
|
| 321 |
+
text_y=0.2,
|
| 322 |
+
text='$p={}$'.format(round(0.001,3)),
|
| 323 |
+
fontsize=11,fontcolor='#000000',
|
| 324 |
+
horizontalalignment='center',)
|
| 325 |
+
|
| 326 |
+
|
| 327 |
+
# ## Boxplot with jitter points
|
| 328 |
+
# A box plot, also known as a box-and-whisker plot, is a graphical representation used to display the distribution and summary statistics of a dataset. It provides a concise and visual way to understand the central tendency, spread, and potential outliers in the data.
|
| 329 |
+
|
| 330 |
+
# **Function**: `ov.pl.single_group_boxplot`:
|
| 331 |
+
#
|
| 332 |
+
# - adata (AnnData object): The data object containing the information for plotting.
|
| 333 |
+
# - groupby (str): The variable used for grouping the data
|
| 334 |
+
# - color (str): The variable used for coloring the data points.
|
| 335 |
+
# - type_color_dict (dict): A dictionary mapping group categories to specific colors.
|
| 336 |
+
# - scatter_kwargs (dict): Additional keyword arguments for customizing the scatter plot.
|
| 337 |
+
# - ax (matplotlib.axes.Axes): A pre-existing axes object for plotting (optional). (optional).(optional).
|
| 338 |
+
#
|
| 339 |
+
|
| 340 |
+
# In[21]:
|
| 341 |
+
|
| 342 |
+
|
| 343 |
+
import pandas as pd
|
| 344 |
+
import seaborn as sns
|
| 345 |
+
#sns.set_style('white')
|
| 346 |
+
|
| 347 |
+
ov.pl.single_group_boxplot(adata,groupby='clusters',
|
| 348 |
+
color='Sox_aucell',
|
| 349 |
+
type_color_dict=dict(zip(pd.Categorical(adata.obs['clusters']).categories, adata.uns['clusters_colors'])),
|
| 350 |
+
x_ticks_plot=True,
|
| 351 |
+
figsize=(5,2),
|
| 352 |
+
kruskal_test=True,
|
| 353 |
+
ylabel='Sox_aucell',
|
| 354 |
+
legend_plot=False,
|
| 355 |
+
bbox_to_anchor=(1,1),
|
| 356 |
+
title='Expression',
|
| 357 |
+
scatter_kwargs={'alpha':0.8,'s':10,'marker':'o'},
|
| 358 |
+
point_number=15,
|
| 359 |
+
sort=False,
|
| 360 |
+
save=False,
|
| 361 |
+
)
|
| 362 |
+
plt.grid(False)
|
| 363 |
+
plt.xticks(rotation=90,fontsize=12)
|
| 364 |
+
|
| 365 |
+
|
| 366 |
+
# ## Complexheatmap
|
| 367 |
+
#
|
| 368 |
+
# A complex heatmap, also known as a clustered heatmap, is a data visualization technique used to represent complex relationships and patterns in multivariate data. It combines several elements, including clustering, color mapping, and hierarchical organization, to provide a comprehensive view of data across multiple dimensions.
|
| 369 |
+
|
| 370 |
+
# **Function**: `ov.pl.single_group_boxplot`:
|
| 371 |
+
#
|
| 372 |
+
# - adata (AnnData): Annotated data object containing single-cell RNA-seq data.
|
| 373 |
+
# - groupby (str, optional): Grouping variable for the heatmap. Default is ''.
|
| 374 |
+
# - figsize (tuple, optional): Figure size. Default is (6, 10).
|
| 375 |
+
# - layer (str, optional): Data layer to use. Default is None.
|
| 376 |
+
# - use_raw (bool, optional): Whether to use the raw data. Default is False.
|
| 377 |
+
# - var_names (list or None, optional): List of genes to include in the heatmap. Default is None.
|
| 378 |
+
# - gene_symbols (None, optional): Not used in the function.
|
| 379 |
+
# - standard_scale (str, optional): Method for standardizing values. Options: 'obs', 'var', None. Default is None.
|
| 380 |
+
# - col_color_bars (dict, optional): Dictionary mapping columns types to colors.
|
| 381 |
+
# - col_color_labels (dict, optional): Dictionary mapping column labels to colors.
|
| 382 |
+
# - left_color_bars (dict, optional): Dictionary mapping left types to colors.
|
| 383 |
+
# - left_color_labels (dict, optional): Dictionary mapping left labels to colors.
|
| 384 |
+
# - right_color_bars (dict, optional): Dictionary mapping right types to colors.
|
| 385 |
+
# - right_color_labels (dict, optional): Dictionary mapping right labels to colors.
|
| 386 |
+
# - marker_genes_dict (dict, optional): Dictionary mapping cell types to marker genes.
|
| 387 |
+
# - index_name (str, optional): Name for the index column in the melted DataFrame. Default is ''.
|
| 388 |
+
# - value_name (str, optional): Name for the value column in the melted DataFrame. Default is ''.
|
| 389 |
+
# - cmap (str, optional): Colormap for the heatmap. Default is 'parula'.
|
| 390 |
+
# - xlabel (str, optional): X-axis label. Default is ''.
|
| 391 |
+
# - ylabel (str, optional): Y-axis label. Default is ''.
|
| 392 |
+
# - label (str, optional): Label for the plot. Default is ''.
|
| 393 |
+
# - save (bool, optional): Whether to save the plot. Default is False.
|
| 394 |
+
# - save_pathway (str, optional): File path for saving the plot. Default is ''.
|
| 395 |
+
# - legend_gap (int, optional): Gap between legend items. Default is 7.
|
| 396 |
+
# - legend_hpad (int, optional): Horizontal space between the heatmap and legend, default is 2 [mm].
|
| 397 |
+
# - show (bool, optional): Whether to display the plot. Default is False.
|
| 398 |
+
#
|
| 399 |
+
#
|
| 400 |
+
|
| 401 |
+
# In[22]:
|
| 402 |
+
|
| 403 |
+
|
| 404 |
+
import pandas as pd
|
| 405 |
+
marker_genes_dict = {
|
| 406 |
+
'Sox':['Sox4', 'Sox7', 'Sox18', 'Sox5'],
|
| 407 |
+
}
|
| 408 |
+
|
| 409 |
+
color_dict = {'Sox':'#EFF3D8',}
|
| 410 |
+
|
| 411 |
+
gene_color_dict = {}
|
| 412 |
+
gene_color_dict_black = {}
|
| 413 |
+
for cell_type, genes in marker_genes_dict.items():
|
| 414 |
+
cell_type_color = color_dict.get(cell_type)
|
| 415 |
+
for gene in genes:
|
| 416 |
+
gene_color_dict[gene] = cell_type_color
|
| 417 |
+
gene_color_dict_black[gene] = '#000000'
|
| 418 |
+
|
| 419 |
+
cm = ov.pl.complexheatmap(adata,
|
| 420 |
+
groupby ='clusters',
|
| 421 |
+
figsize =(5,2),
|
| 422 |
+
layer = None,
|
| 423 |
+
use_raw = False,
|
| 424 |
+
standard_scale = 'var',
|
| 425 |
+
col_color_bars = dict(zip(pd.Categorical(adata.obs['clusters']).categories, adata.uns['clusters_colors'])),
|
| 426 |
+
col_color_labels = dict(zip(pd.Categorical(adata.obs['clusters']).categories, adata.uns['clusters_colors'])),
|
| 427 |
+
left_color_bars = color_dict,
|
| 428 |
+
left_color_labels = None,
|
| 429 |
+
right_color_bars = color_dict,
|
| 430 |
+
right_color_labels = gene_color_dict_black,
|
| 431 |
+
marker_genes_dict = marker_genes_dict,
|
| 432 |
+
cmap = 'coolwarm', #parula,jet
|
| 433 |
+
legend_gap = 15,
|
| 434 |
+
legend_hpad = 0,
|
| 435 |
+
left_add_text = True,
|
| 436 |
+
col_split_gap = 2,
|
| 437 |
+
row_split_gap = 1,
|
| 438 |
+
col_height = 6,
|
| 439 |
+
left_height = 4,
|
| 440 |
+
right_height = 6,
|
| 441 |
+
col_split = None,
|
| 442 |
+
row_cluster = False,
|
| 443 |
+
col_cluster = False,
|
| 444 |
+
value_name='Gene',
|
| 445 |
+
xlabel = "Expression of selected genes",
|
| 446 |
+
label = 'Gene Expression',
|
| 447 |
+
save = True,
|
| 448 |
+
show = False,
|
| 449 |
+
legend = False,
|
| 450 |
+
plot_legend = False,
|
| 451 |
+
#save_pathway = "complexheatmap.png",
|
| 452 |
+
)
|
| 453 |
+
|
| 454 |
+
|
| 455 |
+
# ## Marker gene plot
|
| 456 |
+
#
|
| 457 |
+
# In single-cell analysis, a marker gene heatmap is a powerful visualization tool that helps researchers to understand the expression patterns of specific marker genes across different cell populations. Here we provide `ov.pl.marker_heatmap` for visualizing the patterns of marker genes.
|
| 458 |
+
|
| 459 |
+
# We first preprocess the data and define the dictionary of cell type and marker gene.
|
| 460 |
+
# **Please ensure that each gene in the dictionary appears only once** (i.e. different cells cannot have the same marker gene, otherwise an error will be reported).
|
| 461 |
+
|
| 462 |
+
# In[23]:
|
| 463 |
+
|
| 464 |
+
|
| 465 |
+
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
|
| 466 |
+
|
| 467 |
+
marker_genes_dict = {'Granule immature': ['Sepw1', 'Camk2b', 'Cnih2'],
|
| 468 |
+
'Radial Glia-like': ['Dbi', 'Fabp7', 'Aldoc'],
|
| 469 |
+
'Granule mature': ['Malat1', 'Rasl10a', 'Ppp3ca'],
|
| 470 |
+
'Neuroblast': ['Igfbpl1', 'Tubb2b', 'Tubb5'],
|
| 471 |
+
'Microglia': ['Lgmn', 'C1qa', 'C1qb'],
|
| 472 |
+
'Cajal Retzius': ['Diablo', 'Ramp1', 'Stmn1'],
|
| 473 |
+
'OPC': ['Olig1', 'C1ql1', 'Pllp'],
|
| 474 |
+
'Cck-Tox': ['Tshz2', 'Cck', 'Nap1l5'],
|
| 475 |
+
'GABA': ['Gad2', 'Gad1', 'Snhg11'],
|
| 476 |
+
'Endothelial': ['Sparc', 'Myl12a', 'Itm2a'],
|
| 477 |
+
'Astrocytes': ['Apoe', 'Atp1a2'],
|
| 478 |
+
'OL': ['Plp1', 'Mog', 'Mag'],
|
| 479 |
+
'Mossy': ['Arhgdig', 'Camk4'],
|
| 480 |
+
'nIPC': ['Hmgn2', 'Ptma', 'H2afz']}
|
| 481 |
+
|
| 482 |
+
|
| 483 |
+
# **Function**: `ov.pl.marker_heatmap`:
|
| 484 |
+
#
|
| 485 |
+
# - adata: AnnData object
|
| 486 |
+
# Annotated data matrix.
|
| 487 |
+
# - marker_genes_dict: dict
|
| 488 |
+
# A dictionary containing the marker genes for each cell type.
|
| 489 |
+
# - groupby: str
|
| 490 |
+
# The key in adata.obs that will be used for grouping the cells.
|
| 491 |
+
# - color_map: str
|
| 492 |
+
# The color map to use for the value of heatmap.
|
| 493 |
+
# - use_raw: bool
|
| 494 |
+
# Whether to use the raw data of AnnDta object for plotting.
|
| 495 |
+
# - standard_scale: str
|
| 496 |
+
# The standard scale for the heatmap.
|
| 497 |
+
# - expression_cutoff: float
|
| 498 |
+
# The cutoff value for the expression of genes.
|
| 499 |
+
# - bbox_to_anchor: tuple
|
| 500 |
+
# The position of the legend bbox (x, y) in axes coordinates.
|
| 501 |
+
# - figsize: tuple
|
| 502 |
+
# The size of the plot figure in inches (width, height).
|
| 503 |
+
# - spines: bool
|
| 504 |
+
# Whether to show the spines of the plot.
|
| 505 |
+
# - fontsize: int
|
| 506 |
+
# The font size of the text in the plot.
|
| 507 |
+
# - show_rownames: bool
|
| 508 |
+
# Whether to show the row names in the heatmap.
|
| 509 |
+
# - show_colnames: bool
|
| 510 |
+
# Whether to show the column names in the heatmap.
|
| 511 |
+
# - save_pathway: str
|
| 512 |
+
# The file path for saving the plot.
|
| 513 |
+
# - ax: matplotlib.axes.Axes
|
| 514 |
+
# A pre-existing axes object for plotting (optional).
|
| 515 |
+
|
| 516 |
+
# In[24]:
|
| 517 |
+
|
| 518 |
+
|
| 519 |
+
ov.pl.marker_heatmap(
|
| 520 |
+
adata,
|
| 521 |
+
marker_genes_dict,
|
| 522 |
+
groupby='clusters',
|
| 523 |
+
color_map="RdBu_r",
|
| 524 |
+
use_raw=False,
|
| 525 |
+
standard_scale="var",
|
| 526 |
+
expression_cutoff=0.0,
|
| 527 |
+
fontsize=12,
|
| 528 |
+
bbox_to_anchor=(7, -2),
|
| 529 |
+
figsize=(8.5,4),
|
| 530 |
+
spines=False,
|
| 531 |
+
show_rownames=False,
|
| 532 |
+
show_colnames=True,
|
| 533 |
+
)
|
| 534 |
+
|
ovrawm/t_wgcna.txt
ADDED
|
@@ -0,0 +1,252 @@
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 1 |
+
#!/usr/bin/env python
|
| 2 |
+
# coding: utf-8
|
| 3 |
+
|
| 4 |
+
# # WGCNA (Weighted gene co-expression network analysis) analysis
|
| 5 |
+
# Weighted gene co-expression network analysis (WGCNA) is a systems biology approach to characterize gene association patterns between different samples and can be used to identify highly synergistic gene sets and identify candidate biomarker genes or therapeutic targets based on the endogeneity of the gene sets and the association between the gene sets and the phenotype.
|
| 6 |
+
#
|
| 7 |
+
# Paper: [WGCNA: an R package for weighted correlation network analysis](https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-9-559#Sec21)
|
| 8 |
+
#
|
| 9 |
+
# Narges Rezaie, Farilie Reese, Ali Mortazavi, PyWGCNA: a Python package for weighted gene co-expression network analysis, Bioinformatics, Volume 39, Issue 7, July 2023, btad415, https://doi.org/10.1093/bioinformatics/btad415
|
| 10 |
+
#
|
| 11 |
+
# Code: Reproduce by Python. Raw is http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/Rpackages/WGCNA
|
| 12 |
+
#
|
| 13 |
+
# Colab_Reproducibility:https://colab.research.google.com/drive/1EbP-Tq1IwYO9y1_-zzw23XlPbzrxP0og?usp=sharing
|
| 14 |
+
#
|
| 15 |
+
# Here, you will be briefly guided through the basics of how to use omicverse to perform wgcna anlysis. Once you are set
|
| 16 |
+
|
| 17 |
+
# In[1]:
|
| 18 |
+
|
| 19 |
+
|
| 20 |
+
import scanpy as sc
|
| 21 |
+
import omicverse as ov
|
| 22 |
+
import matplotlib.pyplot as plt
|
| 23 |
+
ov.plot_set()
|
| 24 |
+
|
| 25 |
+
|
| 26 |
+
# ## Load the data
|
| 27 |
+
# The analysis is based on the in-built WGCNA tutorial data. All the data can be download from https://github.com/mortazavilab/PyWGCNA/tree/main/tutorials/5xFAD_paper
|
| 28 |
+
|
| 29 |
+
# In[2]:
|
| 30 |
+
|
| 31 |
+
|
| 32 |
+
import pandas as pd
|
| 33 |
+
data=ov.utils.read('data/5xFAD_paper/expressionList.csv',
|
| 34 |
+
index_col=0)
|
| 35 |
+
data.head()
|
| 36 |
+
|
| 37 |
+
|
| 38 |
+
# In[3]:
|
| 39 |
+
|
| 40 |
+
|
| 41 |
+
from statsmodels import robust #import package
|
| 42 |
+
gene_mad=data.apply(robust.mad) #use function to calculate MAD
|
| 43 |
+
data=data.T
|
| 44 |
+
data=data.loc[gene_mad.sort_values(ascending=False).index[:2000]]
|
| 45 |
+
data.head()
|
| 46 |
+
|
| 47 |
+
|
| 48 |
+
# In[5]:
|
| 49 |
+
|
| 50 |
+
|
| 51 |
+
#import PyWGCNA
|
| 52 |
+
pyWGCNA_5xFAD = ov.bulk.pyWGCNA(name='5xFAD_2k',
|
| 53 |
+
species='mus musculus',
|
| 54 |
+
geneExp=data.T,
|
| 55 |
+
outputPath='',
|
| 56 |
+
save=True)
|
| 57 |
+
pyWGCNA_5xFAD.geneExpr.to_df().head(5)
|
| 58 |
+
|
| 59 |
+
|
| 60 |
+
# ## Pre-processing workflow
|
| 61 |
+
#
|
| 62 |
+
# PyWGCNA allows you to easily preproces the data including removing genes with too many missing values or lowly-expressed genes across samples (by default we suggest to remove genes without that are expressed below 1 TPM) and removing samples with too many missing values. Keep in your mind that these options can be adjusted by changing `TPMcutoff` and `cut`
|
| 63 |
+
|
| 64 |
+
# In[6]:
|
| 65 |
+
|
| 66 |
+
|
| 67 |
+
pyWGCNA_5xFAD.preprocess()
|
| 68 |
+
|
| 69 |
+
|
| 70 |
+
# ## Construction of the gene network and identification of modules
|
| 71 |
+
#
|
| 72 |
+
# PyWGCNA compresses all the steps of network construction and module detection in one function called `findModules` which performs the following steps:
|
| 73 |
+
# 1. Choosing the soft-thresholding power: analysis of network topology
|
| 74 |
+
# 2. Co-expression similarity and adjacency
|
| 75 |
+
# 3. Topological Overlap Matrix (TOM)
|
| 76 |
+
# 4. Clustering using TOM
|
| 77 |
+
# 5. Merging of modules whose expression profiles are very similar
|
| 78 |
+
#
|
| 79 |
+
# In this tutorial, we will perform the analysis step by step.
|
| 80 |
+
|
| 81 |
+
# In[7]:
|
| 82 |
+
|
| 83 |
+
|
| 84 |
+
pyWGCNA_5xFAD.calculate_soft_threshold()
|
| 85 |
+
|
| 86 |
+
|
| 87 |
+
# In[8]:
|
| 88 |
+
|
| 89 |
+
|
| 90 |
+
pyWGCNA_5xFAD.calculating_adjacency_matrix()
|
| 91 |
+
|
| 92 |
+
|
| 93 |
+
# In[9]:
|
| 94 |
+
|
| 95 |
+
|
| 96 |
+
pyWGCNA_5xFAD.calculating_TOM_similarity_matrix()
|
| 97 |
+
|
| 98 |
+
|
| 99 |
+
# ## Building a network of co-expressions
|
| 100 |
+
#
|
| 101 |
+
# We use the dynamicTree to build the co-expressions module basing TOM matrix
|
| 102 |
+
|
| 103 |
+
# In[10]:
|
| 104 |
+
|
| 105 |
+
|
| 106 |
+
pyWGCNA_5xFAD.calculate_geneTree()
|
| 107 |
+
pyWGCNA_5xFAD.calculate_dynamicMods(kwargs_function={'cutreeHybrid': {'deepSplit': 2, 'pamRespectsDendro': False}})
|
| 108 |
+
pyWGCNA_5xFAD.calculate_gene_module(kwargs_function={'moduleEigengenes': {'softPower': 8}})
|
| 109 |
+
|
| 110 |
+
|
| 111 |
+
# In[11]:
|
| 112 |
+
|
| 113 |
+
|
| 114 |
+
pyWGCNA_5xFAD.plot_matrix(save=False)
|
| 115 |
+
|
| 116 |
+
|
| 117 |
+
# ## Saving and loading your PyWGCNA
|
| 118 |
+
# You can save or load your PyWGCNA object with the `saveWGCNA()` or `readWGCNA()` functions respectively.
|
| 119 |
+
|
| 120 |
+
# In[12]:
|
| 121 |
+
|
| 122 |
+
|
| 123 |
+
pyWGCNA_5xFAD.saveWGCNA()
|
| 124 |
+
|
| 125 |
+
|
| 126 |
+
# In[2]:
|
| 127 |
+
|
| 128 |
+
|
| 129 |
+
pyWGCNA_5xFAD=ov.bulk.readWGCNA('5xFAD_2k.p')
|
| 130 |
+
|
| 131 |
+
|
| 132 |
+
# In[14]:
|
| 133 |
+
|
| 134 |
+
|
| 135 |
+
pyWGCNA_5xFAD.mol.head()
|
| 136 |
+
|
| 137 |
+
|
| 138 |
+
# In[15]:
|
| 139 |
+
|
| 140 |
+
|
| 141 |
+
pyWGCNA_5xFAD.datExpr.var.head()
|
| 142 |
+
|
| 143 |
+
|
| 144 |
+
# ## Sub co-expression module
|
| 145 |
+
#
|
| 146 |
+
# Sometimes we are interested in a gene, or a module of a pathway, and we need to extract the sub-modules of the gene for analysis and mapping. For example, we have selected two modules, 6 and 12, as sub-modules for analysis
|
| 147 |
+
|
| 148 |
+
# In[13]:
|
| 149 |
+
|
| 150 |
+
|
| 151 |
+
sub_mol=pyWGCNA_5xFAD.get_sub_module(['gold','lightgreen'],
|
| 152 |
+
mod_type='module_color')
|
| 153 |
+
sub_mol.head(),sub_mol.shape
|
| 154 |
+
|
| 155 |
+
|
| 156 |
+
# We found a total of 151 genes for 'gold' and 'lightgreen'. Next, we used the scale-free network constructed earlier, with the threshold set to 0.95, to construct a gene correlation network graph for modules 'gold' and 'lightgreen'
|
| 157 |
+
|
| 158 |
+
# In[17]:
|
| 159 |
+
|
| 160 |
+
|
| 161 |
+
G_sub=pyWGCNA_5xFAD.get_sub_network(mod_list=['lightgreen'],
|
| 162 |
+
mod_type='module_color',correlation_threshold=0.2)
|
| 163 |
+
G_sub
|
| 164 |
+
|
| 165 |
+
|
| 166 |
+
# In[18]:
|
| 167 |
+
|
| 168 |
+
|
| 169 |
+
len(G_sub.edges())
|
| 170 |
+
|
| 171 |
+
|
| 172 |
+
# pyWGCNA provides a simple visualisation function `plot_sub_network` to visualise the gene-free network of our interest.
|
| 173 |
+
|
| 174 |
+
# In[19]:
|
| 175 |
+
|
| 176 |
+
|
| 177 |
+
pyWGCNA_5xFAD.plot_sub_network(['gold','lightgreen'],pos_type='kamada_kawai',pos_scale=10,pos_dim=2,
|
| 178 |
+
figsize=(8,8),node_size=10,label_fontsize=8,correlation_threshold=0.2,
|
| 179 |
+
label_bbox={"ec": "white", "fc": "white", "alpha": 0.6})
|
| 180 |
+
|
| 181 |
+
|
| 182 |
+
# We also can merge two previous steps by calling `runWGCNA()` function.
|
| 183 |
+
#
|
| 184 |
+
# ## Updating sample information and assiging color to them for dowstream analysis
|
| 185 |
+
|
| 186 |
+
# In[3]:
|
| 187 |
+
|
| 188 |
+
|
| 189 |
+
pyWGCNA_5xFAD.updateSampleInfo(path='data/5xFAD_paper/sampleInfo.csv', sep=',')
|
| 190 |
+
|
| 191 |
+
# add color for metadata
|
| 192 |
+
pyWGCNA_5xFAD.setMetadataColor('Sex', {'Female': 'green',
|
| 193 |
+
'Male': 'yellow'})
|
| 194 |
+
pyWGCNA_5xFAD.setMetadataColor('Genotype', {'5xFADWT': 'darkviolet',
|
| 195 |
+
'5xFADHEMI': 'deeppink'})
|
| 196 |
+
pyWGCNA_5xFAD.setMetadataColor('Age', {'4mon': 'thistle',
|
| 197 |
+
'8mon': 'plum',
|
| 198 |
+
'12mon': 'violet',
|
| 199 |
+
'18mon': 'purple'})
|
| 200 |
+
pyWGCNA_5xFAD.setMetadataColor('Tissue', {'Hippocampus': 'red',
|
| 201 |
+
'Cortex': 'blue'})
|
| 202 |
+
|
| 203 |
+
|
| 204 |
+
# **note**: For doing downstream analysis, we keep aside the Gray modules which is the collection of genes that could not be assigned to any other module.
|
| 205 |
+
#
|
| 206 |
+
# ## Relating modules to external information and identifying important genes
|
| 207 |
+
# PyWGCNA gather some important analysis after identifying modules in `analyseWGCNA()` function including:
|
| 208 |
+
#
|
| 209 |
+
# 1. Quantifying module–trait relationship
|
| 210 |
+
# 2. Gene relationship to trait and modules
|
| 211 |
+
#
|
| 212 |
+
# Keep in your mind before you start analysis to add any sample or gene information.
|
| 213 |
+
#
|
| 214 |
+
# For showing module relationship heatmap, PyWGCNA needs user to choose and set colors from [Matplotlib colors](https://matplotlib.org/stable/gallery/color/named_colors.html) for metadata by using `setMetadataColor()` function.
|
| 215 |
+
#
|
| 216 |
+
# You also can select which data trait in which order you wish to show in module eigengene heatmap
|
| 217 |
+
|
| 218 |
+
# In[4]:
|
| 219 |
+
|
| 220 |
+
|
| 221 |
+
pyWGCNA_5xFAD.analyseWGCNA()
|
| 222 |
+
|
| 223 |
+
|
| 224 |
+
# In[5]:
|
| 225 |
+
|
| 226 |
+
|
| 227 |
+
metadata = pyWGCNA_5xFAD.datExpr.obs.columns.tolist()
|
| 228 |
+
|
| 229 |
+
|
| 230 |
+
# In[10]:
|
| 231 |
+
|
| 232 |
+
|
| 233 |
+
pyWGCNA_5xFAD.plotModuleEigenGene('lightgreen', metadata, show=True)
|
| 234 |
+
|
| 235 |
+
|
| 236 |
+
# In[11]:
|
| 237 |
+
|
| 238 |
+
|
| 239 |
+
pyWGCNA_5xFAD.barplotModuleEigenGene('lightgreen', metadata, show=True)
|
| 240 |
+
|
| 241 |
+
|
| 242 |
+
# ## Finding hub genes for each modules
|
| 243 |
+
#
|
| 244 |
+
# you can also ask about hub genes in each modules based on their connectivity by using `top_n_hub_genes()` function.
|
| 245 |
+
#
|
| 246 |
+
# It will give you dataframe sorted by connectivity with additional gene information you have in your expression data.
|
| 247 |
+
|
| 248 |
+
# In[12]:
|
| 249 |
+
|
| 250 |
+
|
| 251 |
+
pyWGCNA_5xFAD.top_n_hub_genes(moduleName="lightgreen", n=10)
|
| 252 |
+
|